|
|
Costantino Salerno |
|
|
Appunti di Biochimica Clinica |
|
|
8. PROTEINE PLASMATICHE
Le proteine plasmatiche sono un insieme di macromolecole notevolmente eterogenee per quanto riguarda le caratteristiche fisico-chimiche, l’origine e la funzione. Esse formano una soluzione relativamente stabile e possono essere distinte, sulla base della solubilità, in albumine (solubili in acqua) e globuline (solubili in soluzioni saline diluite). La diminuzione della quota albuminica al di sotto del 50% e l’aumento contemporaneo delle frazioni globuliniche comportano una progressiva diminuzione della stabilità colloidale del plasma.
Le proteine plasmatiche sono sintetizzate in netta prevalenza dal fegato. Fra le proteine di origine extraepatica, vi sono le immunoglobuline (sintetizzate dalle plasmacellule; vedi Par. 8.6), alcune componenti del complemento (prodotte dai macrofagi; vedi Par. 8.5), alcune apolipoproteine (formate dall’intestino; vedi Par. 3), gli ormoni proteici (prodotti da specifiche cellule a funzione endocrina; vedi Par. 10) e numerosi enzimi (derivanti dalle cellule dei diversi tessuti per alterazioni reversibili della membrana cellulare o a seguito di processi di necrosi; vedi Par. 9). Le proteine plasmatiche vengono degradate principalmente a livello epatico e reticoloendoteliale, in quanto le perdite fisiologiche renali (vedi Par. 11.2), cutanee e gastrointestinali sono in genere limitate. In alcuni casi (es. aptoglobina, transferrina; vedi Par. 8.1), la quantità catabolizzata giornalmente è relativamente costante. In altri casi (es. fibrinogeno; vedi Par. 7.3.1f), la velocità con cui procede l’eliminazione della proteina è proporzionale alla sua concentrazione plasmatica. Altre volte (es. albumina, IgG1 e IgG2, vedi Par. 8.1.1 e Par. 8.6.1a) il processo catabolico può soggiacere a più fini meccanismi omeostatici tendenti a compensare eventuali squilibri nella concentrazione plasmatica.
Le proteine plasmatiche possono essere classificate in vario modo, tenendo conto delle loro funzioni, delle loro proprietà fisico-chimiche, del loro significato diagnostico o di altri fattori.
8.1. PROTEINE DI TRASPORTO O DI LEGAME
Numerose proteine, in parte ancora poco note, possono essere incluse in questo gruppo. Molte di esse svolgono anche altre funzioni o mostrano un comportamento del tipo "risposta di fase acuta" (vedi Par. 8.2) e sono così classificabili altrove. Ci limiteremo perciò ad esaminare solo alcune di queste proteine.
8.1.1. Albumina
L’albumina è costituita da una singola catena
polipeptidica di 66 kDa. Essa presenta 17 ponti disolfuro
intramolecolari, che portano al ripiegamento della proteina in
una struttura elissoidale distinta in tre regioni separabili fra
loro mediante una proteolisi parziale. Esistono numerose varianti
genetiche dell’albumina elettroforeticamente distinguibili.
La presenza di albumina con una anomala mobilità elettroforetica
è indicata con il termine di alloalbuminemia, mentre la
contemporanea presenza di due frazioni elettroforeticamente
distinte prende il nome di bisalbuminemia
![]() .
.
La proteina è sintetizzata dal fegato ed ha un’emivita di circa 20 giorni. Solo il 30-40% dell’albumina è contenuto nel plasma; la rimanente quota si trova nel liquido interstiziale dei vari tessuti (principalmente nel muscolo, nella cute e nell’intestino). Circa il 5% dell’albumina plasmatica lascia il sangue ogni ora e una pari quantità rientra nel torrente circolatorio con la linfa del dotto toracico. La concentrazione dell’albumina nel siero è di 35-55 g/L (0,5-0,8 mmol/L) nell’adulto. Valori leggermente più bassi si osservano nei neonati e nei bambini. Nei soggetti giacenti a lungo a letto, la concentrazione dell’albumina nel plasma diminuisce di circa 5 g/L.
Questa proteina, che costituisce da sola il 50-60% di tutte le proteine plasmatiche, è responsabile (per il 75-80%) dell’effetto colloido-osmotico del plasma. Essa svolge inoltre un’importante funzione di trasporto veicolando numerose sostanze (bilirubina, acidi grassi liberi, ormoni, vitamine, farmaci, etc.). Infine, l’albumina è utilizzata dai tessuti come fonte di aminoacidi per il fabbisogno nutrizionale delle cellule.
Un’iperalbuminemia si può riscontrare in quegli stati
morbosi (diarrea e vomito persistenti, ileo occlusivo, ustioni
estese, coma diabetico, morbo di Addison, etc.) che comportano
una disidratazione e un conseguente aumento della concentrazione
delle componenti ematiche non diffusibili. Una diminuzione della
concentrazione dell’albumina nel siero è generalmente di più
frequente riscontro e può essere dovuta ad emodiluizione (conseguente
ad uno sbilanciamento elettrolitico) o un alterato rapporto fra
sintesi e perdita di questa frazione proteica. Un deficit
congenito nella sintesi di albumina, peraltro spesso
asintomatico, si osserva nei rari casi di analbuminemia. La
sintesi dell’albumina può inoltre diminuire a seguito di un
insufficiente apporto alimentare (grave denutrizione
![]() , sindromi da malassorbimento, stati cachettici), di una grave e prolungata
insufficienza epatocellulare (epatopatie acute e croniche di
varia natura) o nel contesto di una "risposta di fase acuta" (vedi
Par. 8.2).
Un aumentato catabolismo proteico, che comporta una diminuzione
della concentrazione dell’albumina nel siero, si può
riscontrare negli stati febbrili di lunga durata, nella tireotossicosi (vedi
Par. 10.4), nella sindrome di Cushing (vedi
Par. 10.5.1d) e nelle
neoplasie. Una eccessiva perdita di albumina si può avere infine
nella sindrome nefrosica
, sindromi da malassorbimento, stati cachettici), di una grave e prolungata
insufficienza epatocellulare (epatopatie acute e croniche di
varia natura) o nel contesto di una "risposta di fase acuta" (vedi
Par. 8.2).
Un aumentato catabolismo proteico, che comporta una diminuzione
della concentrazione dell’albumina nel siero, si può
riscontrare negli stati febbrili di lunga durata, nella tireotossicosi (vedi
Par. 10.4), nella sindrome di Cushing (vedi
Par. 10.5.1d) e nelle
neoplasie. Una eccessiva perdita di albumina si può avere infine
nella sindrome nefrosica
![]() ,
nelle enteropatie proteinodisperdenti, nelle lesioni essudative
cutanee e nelle emorragie di varia natura. L’ipoalbuminemia
che si osserva in gravidanza è imputabile sia alla ritenzione
idrica sia all’aumentata utilizzazione delle proteine da
parte del feto.
,
nelle enteropatie proteinodisperdenti, nelle lesioni essudative
cutanee e nelle emorragie di varia natura. L’ipoalbuminemia
che si osserva in gravidanza è imputabile sia alla ritenzione
idrica sia all’aumentata utilizzazione delle proteine da
parte del feto.
8.1.2. Prealbumina
La
prealbumina (o transtiretina) è un tetramero
formato dall’associazione di quattro subunità identiche di
14 kDa ciascuna. Essa deve il suo nome al fatto di avere una
mobilità elettroforetica di poco maggiore rispetto all’albumina
quando viene analizzata con le metodiche di comune impiego in
chimica clinica. Questa proteina di origine epatica ha nel siero
una concentrazione di 100-400 mg/L (1,8-7,2 µmol/L)
ed un’emivita molto breve (circa 2 giorni). La prealbumina
concorre a veicolare la tiroxina nel sangue
![]() . Essa forma inoltre
dei complessi isomolecolari con la "proteina legante il retinolo",
contribuendo indirettamente al trasporto ematico della vitamina A.
Mutazioni del gene codificante per la prealbumina, che danno
luogo a singole sostituzioni nella sequenza aminoacidica, possono
essere causa dell’amiloidosi ereditaria sistemica
. Essa forma inoltre
dei complessi isomolecolari con la "proteina legante il retinolo",
contribuendo indirettamente al trasporto ematico della vitamina A.
Mutazioni del gene codificante per la prealbumina, che danno
luogo a singole sostituzioni nella sequenza aminoacidica, possono
essere causa dell’amiloidosi ereditaria sistemica
![]() .
.
La concentrazione ematica della prealbumina risente prontamente di una riduzione della capacità proteinosintetica del fegato, osservabile negli stati di malnutrizione o nel corso di malattie epatiche (nell’epatite virale si assiste, per esempio, ad un precocissimo abbassamento della concentrazione della prealbumina, eventualmente seguito da un progressivo recupero, che è da intendersi come un indice prognostico favorevole per la malattia). Questa proteina si comporta, inoltre, come un "reattante negativo della fase acuta" (vedi Par. 8.2). La concentrazione della prealbumina nel siero aumenta nella sindrome nefrosica e nelle ultime settimane di gravidanza.
8.1.3. Aptoglobina
L’aptoglobina è una glicoproteina sintetizzata dal fegato e costituita da due catene a, poste sotto il controllo di tre alleli denominati α1F, α1S e α2, e da due catene β, sintetizzate da un unico gene. Poiché gli alleli α1F e α1S non sono distinguibili con i comuni metodi di analisi elettroforetica a pH alcalino, i fenotipi presenti nella popolazione sono comunemente classificati come Hp 1-1 (α12β2), Hp 1-2 (α12β2, α1α2β2 e α22β2) e Hp 2-2 (α22β2). Il fenotipo Hp 1-1 presenta una singola molecola tetramerica di 85 kDa che si evidenzia come una unica banda elettroforetica. Il fenotipo Hp 2-2 tende a polimerizzare e dà luogo ad una serie di bande elettroforetiche in posizione più catodica. Il fenotipo Hp 1-2 presenta un quadro elettroforetico che è la risultante dei precedenti. In Europa e nella popolazione americana di origine europea il fenotipo Hp 1-1 è presente nel 13-21% dei casi, il fenotipo 1-2 nel 47-59% dei casi e il fenotipo Hp 2-2 nel 28-36% dei casi. Il fenotipo Hp 1-1 è più comune in Africa mentre il fenotipo Hp 2-2 predomina in Asia. La concentrazione dell’aptoglobina nel siero è di 0,6-2,7 g/L (7-32 µmol/L, come molecola tetramerica) e la sua emivita è di 1-3 giorni. L’aptoglobina è molto bassa o assente nel siero dei neonati e raggiunge valori prossimi a quelli dell’adulto al quarto mese di vita.
L’aptoglobina lega in rapporto stechiometrico e veicola l’emoglobina, derivante da un’eventuale emolisi (intravasale o extravasale), nei distretti del sistema reticolo-endoteliale dove viene catabolizzata. L’aptoglobina diminuisce tutte le volte che vi è un’eccessiva distruzione di emazie circolanti. Ciò costituisce una delle più sensibili prove di laboratorio per documentare stati iperemolitici anche lievi e clinicamente inapparenti. I fenotipi Hp 1-2 e 2-2, ad elevato peso molecolare, possono aumentare in corso di una sindrome nefrosica. La concentrazione dell’aptoglobina nel siero aumenta, inoltre, nella fase acuta di processi infiammatori e reattivi causati da lesioni tessutali di varia natura, in quanto questa molecola rientra nel gruppo delle "proteine della fase acuta" (vedi Par. 8.2). La simultanea presenza di un processo infiammatorio e di un’emolisi può perciò dar luogo sia ad un aumento che ad una diminuzione o non indurre alcun cambiamento nel livello dell’aptoglobina.
8.1.4. Emopessina
L’emopessina (o
β1B-glicoproteina) è una glicoproteina
di 57 kDa prodotta dal fegato e presente nel siero alla
concentrazione di 0,5-1,1 g/L (8,8-19 µmol/L)
con un’emivita di poco più di una settimana. Questa
proteina lega il gruppo eminico libero (uno per molecola) che si
distacca dall’emoglobina a seguito della sua ossidazione a
metaemoglobina
![]() . Il
complesso eme-emopessina viene incorporato nelle cellule epatiche
e quindi degradato.
. Il
complesso eme-emopessina viene incorporato nelle cellule epatiche
e quindi degradato.
L’emopessina sierica diminuisce nelle anemie talassemiche e drepanocitiche (vedi Par. 7.2.1), così come in tutte le altre situazioni morbose caratterizzate da emolisi intravasale (per esempio, è particolarmente utile per seguire il danno sulle emazie causato dalle protesi valvolari intracardiache). A differenza dell’aptoglobina, questa proteina non presenta varianti genetiche ed è quindi più facilmente quantizzabile. Inoltre aumenta in maniera meno spiccata nel corso di una "risposta di fase acuta" ed è perciò meno influenzabile da altre cause patologiche concomitanti. Al di fuori delle malattie con emolisi intravasale, l’emopessina diminuisce nel siero nelle gravi epatopatie carenziali (kwashiorkor) e, talora, nella pancreatite acuta emorragica e nella porfiria (vedi Par. 5.1). L’emopessina nel siero può aumentare nel diabete mellito (vedi Par. 2.1a) , nella distrofia muscolare e delle neoplasie a rapida crescita.
8.1.5. Transferrina
La transferrina è una glicoproteina contenente acido sialico e costituita da una singola catena polipeptidica di 76 kDa. Sono state descritte numerose varianti genetiche distinguibili elettroforeticamente. La molecola è sintetizzata in numerosi tessuti, ma in gran parte origina dal fegato. Circa il 50-60% della trasferrina si trova nel fluido extracellulare (compresi il liquor e la linfa). E’ presente inoltre anche nei fluidi edematosi. E’ presente nel siero ad una concentrazione di 1,9-3,1 g/L (25-40 µmol/L) ed ha una emivita di circa 8 giorni.
La transferrina ha due siti ad alta affinità per il ferro che
trasporta dai tessuti che lo liberano (reticolo endotelio,
intestino, tessuti parenchimatosi) ai tessuti che ne hanno
bisogno (principalmente il tessuto eritropoietico e la placenta)
e che presentano una glicoproteina recettoriale specifica
![]() . Normalmente, la
transferrina è saturata per solo un terzo dal ferro.
. Normalmente, la
transferrina è saturata per solo un terzo dal ferro.
La transferrina diminuisce nel siero quando vi è un
sovraccarico di ferro, mentre aumenta quando il ferro è carente.
La transferrina del siero è inoltre bassa in caso di
malnutrizione o di grave insufficienza epatica, nelle enteropatie
proteinodisperdenti, nella sindrome nefrosica
![]() , negli stati
morbosi che determinano una aumentata emolisi, nell’infarto
del miocardio, nelle neoplasie maligne e nei processi
infiammatori di varia etiologia (comportandosi così come un
"reattante negativo della fase acuta"; vedi Par. 8.2). La tranferrina
è assente nei rari casi di atransferrinemia congenita. Un
aumento della transferrinemia si osserva durante la gravidanza o
l’uso di contraccetivi contenenti estrogeni, anche quando
non vi è una carenza di ferro, per un effetto diretto degli
estrogeni sulla trascrizione del mRNA specifico.
, negli stati
morbosi che determinano una aumentata emolisi, nell’infarto
del miocardio, nelle neoplasie maligne e nei processi
infiammatori di varia etiologia (comportandosi così come un
"reattante negativo della fase acuta"; vedi Par. 8.2). La tranferrina
è assente nei rari casi di atransferrinemia congenita. Un
aumento della transferrinemia si osserva durante la gravidanza o
l’uso di contraccetivi contenenti estrogeni, anche quando
non vi è una carenza di ferro, per un effetto diretto degli
estrogeni sulla trascrizione del mRNA specifico.
8.1.6. Ferritina
La ferritina è una grossa macromolecola costituita da 24 subunità disposte geometricamente a formare un involucro sferico di circa 480 kDa (apoferritina) contenente al suo interno fino a 4500 molecole di idrossifosfato ferrico colloidale. La parte centrale della macromolecola è in comunicazione con l’esterno mediante sei canali attraversanti a tutto spessore il guscio proteico. La ferritina dei diversi tessuti contiene, in proporzioni variabili, due tipi di subunità: la subunità L di 19 kDa, che predomina nella più basica ferritina epatica e splenica, e la subunità H di 21 kDa, maggiormente presente nella più acida ferritina del cuore, rene e placenta.
Per indicare queste diverse ferritine è stato adottato il
termine di "isoferritine", in analogia con la
terminologia usata da tempo per gli enzimi (vedi
Par. 9.1). I quadri isoferritinici tessutali, in particolare quello cardiaco, si
modificano in condizioni di sovraccarico di ferro in quanto, per
un aumento della sintesi delle subunità L, tendono a scomparire
in tali condizioni le frazioni più acide. In molti tumori,
invece, e in particolare nelle neoplasie epatiche si osserva un
aumento delle isoferritine più acide (isoferritine carcinofetali).
La ferritina è contenuta in gran parte nel fegato, nel midollo
osseo e nel muscolo scheletrico, ove è presente nelle cellule sia parinchematose che reticoloendoteliali, fungendo da deposito
intracellulare del ferro; a sua volta, il ferro presente nella cellula è capace di modulare
la sintesi della ferritina
![]() .
.
La ferritina presente nel siero ha un contenuto di ferro nettamente inferiore a quello delle ferritine tessutali, è glicosilata e si comporta come la più basica delle isoferritine umane. La ferritina sierica potrebbe derivare da una lisi cellulare o essere espressamente secreta dalle cellule stesse. In quest’ultimo caso, essa potrebbe avere un ruolo nel regolare la sintesi della transferrina o l’assorbimento del ferro alimentare. La ferritina ha un’emivita di solo 10 minuti ed è captata quasi completamente dagli epatociti. I valori di riferimento per la ferritina sierica in soggetti adulti sono di 90-130 µg/L (nei maschi) e di 30-60 µg/L (nelle femmine). La concentrazione media della ferritina nel sangue del funicolo ombellicale è simile a quella degli adulti (25-200 µg/L); essa aumenta bruscamente nel primo mese di vita (200-600 µg/L) per ridiscendere dopo il sesto mese (7-142 µg/L) e mantenersi costante fino alla pubertà, quando i valori relativi ai maschi iniziano a discostarsi da quelli relativi alle femmine. Dopo la menopausa, la ferritinemia nei soggetti di sesso femminile aumenta avvicinandosi ai valori osservabili nei maschi.
Vi è una stretta relazione fra ferritinemia e quantità di ferro immagazzinato in quanto la concentrazione della ferritina nel plasma aumenta all’aumentare delle riserve in ferro dell’organismo (ogni mg di ferritina presente in un litro di plasma corrisponde ad un accumulo di 8 mg di ferro). Tuttavia, quando la ferritinemia è troppo elevata ( > 4000 µg/L), questa relazione diviene meno precisa a causa di un maggior rilascio di ferritina nel plasma da parte delle cellule danneggiate dall’eccessivo accumulo di ferro o di una saturazione dei meccanismi coinvolti nella secrezione della ferritina plasmatica. Un aumento della ferritinemia (indipendentemente dall’aumento dei depositi di ferro) è osservabile in casi di necrosi acuta delle cellule epatiche, nel corso di malattie neoplastiche (epatoma, carcinoma polmonare, leucemia) o nella risposta di fase acuta (vedi Par. 8.2).
8.1.7. Ceruloplasmina
La ceruloplasmina è una α2-glicoproteina di origine epatica contenente 6 atomi di rame per molecola. La proteina è costituita da una singola catena aminoacidica di 150 kDa ed ha un intenso colore blu, tale da impartire al siero una tinta verdastra quando il livello dell’albumina è notevolmente ridotto. E’ presente nel siero ad una concentrazione di 150-600 mg/L (1-4 µmol/L). La concentrazione della ceruloplasmina è più bassa nel siero dei neonati (70-150 mg/L) e raggiunge valori prossimi a quelli dell’adulto al terzo-sesto mese di vita.
La proteina presenta una attività enzimatica, catalizzando l’ossidazione del ferro(II) da parte dell’ossigeno atmosferico (ferrossidasi, EC 1.16.3.1):
![]()
La reazione è relativamente aspecifica in quanto il ferro(II) può essere sostituito con numerosi altri agenti riducenti (benzidina, p-fenilenediamina, N,N-dimetilfenilenediamina, o-dianisidina), utili per una rilevazione colorimetrica dell’attività enzimatica.
La cerulopasmina trasporta circa il 90% del rame contenuto nel plasma. Studi condotti utilizzando il rame radioattivo hanno dimostrato che questo viene rapidamente incorporato nella ceruloplasmina nelle prime ore successive alla sua somministrazione endovena. In seguito la sua incorporazione nella ceruloplasmina prosegue più lentamente, in modo tale che il 5-10% della dose iniettata viene alla fine incorporato in circa una settimana.
La ceruloplasmina sierica è notevolmente ridotta nei soggetti
affetti dal morbo di Wilson
![]() . La ceruloplasmina sierica può diminuire
anche in conseguenza di un’insufficienza epatica acuta di
varia origine. Il livello di ceruloplasmina è inoltre
influenzabile da differenti fattori genetici. La trasmissione
ereditaria dell’ipoceruloplasminemia è di tipo dominante e non
comporta la comparsa di segni clinici evidenti. La concentrazione
della ceruloplasmina aumenta del 100% nell’ultimo trimestre
di gravidanza o a seguito della somministrazione di
contraccettivi orali. La ceruloplasmina si comporta infine come
una "proteina della fase acuta", aumentando di circa il
50% nel corso delle infezioni acute (vedi Par. 8.2).
. La ceruloplasmina sierica può diminuire
anche in conseguenza di un’insufficienza epatica acuta di
varia origine. Il livello di ceruloplasmina è inoltre
influenzabile da differenti fattori genetici. La trasmissione
ereditaria dell’ipoceruloplasminemia è di tipo dominante e non
comporta la comparsa di segni clinici evidenti. La concentrazione
della ceruloplasmina aumenta del 100% nell’ultimo trimestre
di gravidanza o a seguito della somministrazione di
contraccettivi orali. La ceruloplasmina si comporta infine come
una "proteina della fase acuta", aumentando di circa il
50% nel corso delle infezioni acute (vedi Par. 8.2).
8.2. PROTEINE DELLA FASE ACUTA
La "risposta di fase acuta" è una reazione sistemica ad una varietà di stimoli capaci di causare un danno tessutale culminante in un processo di necrosi cellulare. Essa è caratterizzata da numerose modificazioni metaboliche includenti comparsa di febbre, neutrofilia ed aumentata reattività immunitaria. Durante la risposta di fase acuta si ha un’alterazione della concentrazione della maggior parte delle proteine plasmatiche (ad eccezione delle immunoglobuline che, almeno inizialmente, non variano). Alcune proteine (albumina, prealbumina, proteina legante il retinolo, transferrina, ApoA-I, properdina, α2-HS-glicoproteina) diminuiscono di concentrazione durante questa reazione sistemica comportandosi come dei "reattanti negativi della fase acuta". Numerose altre, circa trenta, aumentano in modo variabile e vengono classificate perciò fra le "proteine della fase acuta". Fra esse, alcune aumentano da due a cinque volte (come le componenti C3 e C4 del complemento, l’α1-antitripsina, l’α1-antichimotripsina, l’aptoglobina, la ceruloplasmina, l’orosomucoide e il fibrinogeno) mentre altre aumentano fino ad anche mille volte (la proteina C reattiva e la proteina sierica dell’amiloide A).
Le modificazioni di concentrazione plasmatica delle proteine della fase acuta riflettono una variazione nella loro velocità di sintesi epatica, a sua volta dovuta alla produzione, da parte delle cellule macrofagiche attivate, di opportuni mediatori, principalmente le interleuchine (IL-1b e IL-6) e il fattore di necrosi tumorale (TNF-a). La proteina C reattiva, la proteina sierica dell’amiloide A e l’α1-antichimotripsina sono incrementate già dopo 6-10 ore dall’insulto flogistico e raggiungono un massimo dopo 48-72 ore. L’α1-antitripsina, l’aptoglobina, l’orosomucoide e il fibrinogeno iniziano ad aumentare dopo 24 ore, mostrando un picco fra le 72 e le 96 ore. La ceruloplasmina e le componenti C3 e C4 del complemento rispondono più lentamente, aumentando solo dopo 48-72 ore. Fra i "reattanti negativi della fase acuta", la più rapida è la prealbumina seguita, nell’ordine, dalla transferrina, dall’ApoA-I e dall’albumina. Alla cessazione dello stimolo flogistico, la concentrazione delle proteine ritorna al livello iniziale nel medesimo ordine.
Le variazioni in concentrazione delle proteine plasmatiche nel corso di una risposta di fase acuta possono essere mascherate da altri processi morbosi concomitanti. Così, la concentrazione del C3 può risultare normale per una contemporanea attivazione della via classica o alternativa del complemento (rispettivamente, nel corso di sepsi o di collagenopatie). L’α1-antitripsina può non aumentare per un deficit genetico (presenza del fenotipo MZ) o nel corso di flogosi che interessano i tessuti mesenchimali (per liberazione di enzimi leucocitari che ne accellerano il catabolismo). L’aumento dell’aptoglobina può non essere apparente se è presente un concomitante processo emolitico. L’orosomucoide può risultare normale in presenza di flogosi intestinale, cirrosi o sindrome nefrosica. L’aumento della sintesi del fibrinogeno può essere mascherata da una coagulazione intravasale disseminata o a seguito dell’assunzione di ormoni steroidei. La somministrazione di ormoni steroidei può mascherare anche la diminuzione di alcuni reattanti negativi della fase acuta (albumina, prealbumina, transferrina).
8.2.1. Proteina C reattiva
La proteina C reattiva è una globulina di origine epatica formata da 5 subunità identiche di 23 kDa ciascuna. Deve il nome al fatto che fu inizialmente identificata per la sua capacità di legarsi, in presenza di calcio, al polisaccaride somatico C estratto da colture di pneumococco. La sua concentrazione nel siero di soggetti adulti sani è inferiore a 10 mg/L (90 nmol/L). In condizioni normali, la sintesi della proteina C reattiva, sebbene sia limitata, avviene probabilmente in risposta ad infezioni o danni tessutali di lieve entità, che sono presenti anche in soggetti apparentemente sani (in effetti, nei sieri ottenuti dal cordone ombellicale la concentrazione è inferiore a 0,1 mg/L).
La massima concentrazione, che si raggiunge nella risposta di fase acuta, è proporzionale all’entità del danno tessutale e dipende dalla durata del processo flogistico, piuttosto che dalla velocità iniziale di sintesi della proteina. Cessato lo stimolo, la concentrazione della proteina C reattiva diminuisce rapidamente dimezzandosi in 24 ore dopo un efficace controllo della flogosi.
La proteina C reattiva, oltre che reagire con il polisaccaride C dello pneumococco, forma un legame calcio-dipendente con i fosfatidi della colina (lecitina, lisolecitina, sfingomielina), con altri lipidi, polisaccaridi e peptidopolisaccaridi, contenenti o meno la fosforilcolina, e con polianioni, inclusi gli acidi nucleici, l’eparina e il solfato di destrano. In assenza di ioni calcio, la proteina C reattiva si lega a policationi (tra cui gli istoni, la proteina cationica dei leucociti e la protamina) utilizzando un sito di legame differente, anche se interagente con quello dipendente dal calcio.
La proteina C reattiva, interagendo con le membrane cellulari danneggiate, ma non con quelle indenni, e con componenti di diversi batteri, funghi e parassiti, riconosce rapidamente, prima che si sviluppi una risposta anticorpale, la presenza di un danno tessutale o di un agente infettante. Una volta complessata, questa proteina attiva il complemento attraverso la via classica e facilita così l’eliminazione del materiale a cui si è legata (vedi Par. 8.5).
8.2.2. Proteina sierica dell’amiloide A
La
proteina sierica dell’amiloide A è una
apolipoproteina associata alle HDL3 (vedi
Par. 3), che esiste in
almeno due forme isotipiche di 11 kDa ed è presente nel siero ad
una concentrazione inferiore a 30 mg/L (3 µmol/L).
E’ probabilmente importante per la captazione del
colesterolo da parte del fegato o per lo smaltimento di altro
materiale tossico liposolubile complessato alle HDL. In vitro, è
stata vista sopprimere la risposta anticorpale dipendente dalle
cellule T. Inoltre questa proteina svolge un ruolo importante
nella patogenesi dell’amiloidosi reattiva (secondaria) in
quanto da un suo frammento traggono origine le fibrille proteiche
costituenti l’amiloide A che si deposita nei tessuti, forse
per un difetto nel normale processo degradativo della proteina
sierica
![]() .
.
8.2.3. Orosomucoide
L’orosomucoide o
α1-glicoproteina
acida è una mucoproteina di circa 44 kDa con una funzione
biologica ancora poco conosciuta, presente nel siero alla
concentrazione di 0,55-1,2 g/L (12-27 µmol/L)
![]() . Oltre che nella
risposta di fase acuta, l’orosomucoide aumenta negli itteri
ostruttivi (specialmente di natura angiocolitica) mentre
diminuisce negli itteri epatocellulari, nelle flogosi intestinali
e nella sindrome nefrosica
. Oltre che nella
risposta di fase acuta, l’orosomucoide aumenta negli itteri
ostruttivi (specialmente di natura angiocolitica) mentre
diminuisce negli itteri epatocellulari, nelle flogosi intestinali
e nella sindrome nefrosica
![]() .
.
8.3. INIBITORI DELLE PROTEASI
Le proteine, che agiscono contrastando l’azione degli enzimi proteolitici, prendono il nome di inibitori delle proteasi. Alcuni inibitori delle proteasi (l’α1-antitripsina, l’α1-antichimotripsina, l’α2-antiplasmina, l’antitrombina III, l’inibitore della C1-esterasi, etc.) fanno parte delle serpine. Con questo termine si indica un gruppo di proteine con strette analogie strutturali capaci di inibire le proteasi a serina, cioè quegli enzimi proteolitici che posseggono un residuo di serina al sito attivo. Il sito reattivo delle serpine è situato su un’ansa della catena aminoacidica che è riconosciuta come substrato dall’enzima proteolitico che viene inattivato.
8.3.1. α1-Antitripsina
L’α1-antitripsina è una glicoproteina monomerica di 52 kDa sintetizzata dal fegato e dai macrofagi con un’emivita di circa 5 giorni e presente nel siero alla concentrazione di 2-3 g/L (40-60 µmol/L).
L’α1-antitripsina
inibisce un ampio spettro di proteasi caratterizzate dalla
presenza di un residuo di serina al sito attivo, comprendente l’elastasi
del pancreas e dei neutrofili, la catepsina G dei neutrofili, la
tripsina e la chimitripsina pancreatica, la collagenasi, la
callicreina, l’urochinasi, la renina e numerose proteasi
della cascata coagulativa, del sistema fibrinolitico e del
complememto. In molti casi, queste azioni inibenti, osservabili
in vitro, sono prive di una effettiva importanza fisiopatologica
![]() . La α1-antitripsina costituisce il
principale inibitore dell’elastasi dei neutrofili nelle sedi
non interessate da processi flogistici: nei tessuti infiammati i
granulociti neutrofili stessi garantiscono l’attività di
questa proteasi mediante il simultaneo rilascio di radicali dell’ossigeno
che inattivano l’α1-antitripsina
ossidandola a livello della metionina-358.
. La α1-antitripsina costituisce il
principale inibitore dell’elastasi dei neutrofili nelle sedi
non interessate da processi flogistici: nei tessuti infiammati i
granulociti neutrofili stessi garantiscono l’attività di
questa proteasi mediante il simultaneo rilascio di radicali dell’ossigeno
che inattivano l’α1-antitripsina
ossidandola a livello della metionina-358.
La sintesi dell’α1-antitripsina è controllata da un unico locus genico chiamato PI (proteinase inhibitor) e localizzato sul braccio lungo del cromosoma 14. Il gene presenta un elevato polimorfismo, essendo stati descritti più di 60 alleli codominanti. Il fenotipo più comune appartiene alla famiglia delle varianti M e raccoglie circa il 95% della popolazione. La maggior parte delle varianti genetiche sono associate ad un normale livello di a1-antitripsina nel plasma. Fanno eccezione le varianti I, S e P che sono associate nell’omozigote alla presenza di livelli plasmatici pari, rispettivamente, al 68, 60 e 30% del normale. Il fenotipo omozigote per la variante Z presenta nel plasma meno del 20% dell’α1-antitripsina con una conseguente insufficiente inibizione della elastasi neutrofilica (l’eterozigote MZ ha circa il 60% della proteina plasmatica ed è asintomatico). La carenza della variante Z dell’α1-antitripsina nel plasma è dovuta ad un difetto nella secrezione della proteina da parte degli epatociti, nei quali essa forma delle inclusioni citoplasmatiche caratteristiche. I soggetti omozigoti per la variante Z, a causa della scarsa protezione verso l’elastasi neutrofilica, hanno un notevole rischio di andare incontro, fra la quarta e la quinta decade di vita, ad enfisema polmonare (particolarmente marcato nei lobi inferiori), la cui comparsa è favorita da infezioni broncopolmonari ricorrenti, da agenti inquinanti atmosferici o dal fumo di sigaretta. L’accumulo intraepatico di α1-antitripsina può determinare, d’altro canto, segni biochimici di alterazione epatica in età infantile e l’insorgenza di cirrosi ed epatoma in età adulta.
L’α1-antitripsina si comporta come una "proteina della fase acuta" (vedi Par. 8.2). Essa aumenta, inoltre, nel corso di gravidanza o durante trattamento estroprogestinico.
8.3.2. α1-Antichimotripsina
L’α1-antichimotripsina è una glicoproteina monomerica di 59 kDa presente nel siero ad una concentrazione di 0,2-0,6 g/L (3-10 µmol/L). Nel neonato la concentrazione plasmatica media è di 0,1 g/L (2 µmol/L). E’ un inibitore delle proteasi simili alla chimotripsina, non agendo, invece, sulla tripsina, plasmina e trombina. Volge inoltre un ruolo nella risposta immune, potenziando l’attività anticorpale ed inibendo l’attività citotossica naturale dei linfociti T killer e quella cellulomediata dipendente dagli anticorpi. Si comporta come una "proteina della fase acuta" nel corso di processi infiammatori e necrotici.
8.3.3. α2-Macroglobulina
La α2-macroglobulina è una glicoproteina formata da 4 unità monomeriche di 180 kDa ciascuna, unite a coppia mediante ponti disolfuro. I valori di riferimento nel siero sono compresi fra 1,5-3,5 g/L (2-5 µmol/L). Valori sensibilmente più elevati sono osservabili nella donna, nel bambino di età inferiore ai 4 anni e, specialmente, nel neonato prematuro. La α2-macroglobulina è in grado di inibire numerose proteasi per le quali presenta due siti leganti e che è in grado di intrappolare ostacolandone stericamente il sito attivo senza coinvolgerlo però direttamente. Le proteasi complessate alla α2-macroglobulina non sono perciò in grado di agire su proteine di dimensioni rilevanti, ma possono idrolizzare peptidi di dimensioni modeste (fino a 20 kDa) così come substrati solubili artificiali. Il complesso α2-macroglobulina-proteasi viene quindi allontanato per endocitosi mediata da recettori presenti sugli epatociti e sulle cellule di Kupffer. Mediante questa azione antiproteasica, la α2-macroglobulina contribuisce alla regolazione del processo coagulativo, fibrinolitico ed immunitario. La proteina contribuisce inoltre al trasporto di alcuni ormoni di natura proteica come il somatotropo e l’insulina.
La α2-macroglobulina
aumenta nel corso della gravidanza o a seguito di
somministrazione di contraccetivi orali. La α2-macroglobulina,
inoltre, è più elevata in alcune distrofie congenite come l’atassia
telangectasica
![]() e
la sindrome di Down
e
la sindrome di Down
![]() . Un
aumento nettissimo si osserva nella sindrome nefrosica
. Un
aumento nettissimo si osserva nella sindrome nefrosica
![]() . Altre
condizioni morbose che non di rado si accompagnano ad un aumento
di α2-macroglobulina sono
il diabete mellito, le epatopatie croniche, il reumatismo
articolare acuto, diverse malattie infettive e le ustioni.
. Altre
condizioni morbose che non di rado si accompagnano ad un aumento
di α2-macroglobulina sono
il diabete mellito, le epatopatie croniche, il reumatismo
articolare acuto, diverse malattie infettive e le ustioni.
Bassi valori di α2-macroglobulina
si riscontrano nell’artrite reumatoide ![]() (cosa che la esclude dal novero delle
"proteine della fase acuta") e nell’eclampsia
gravidica
(cosa che la esclude dal novero delle
"proteine della fase acuta") e nell’eclampsia
gravidica
![]() .
.
8.4. MICROGLOBULINE
Questo gruppo comprende numerose globuline con massa molecolare inferiore a 40 kDa, elettroforeticamente eterogenee e di significato spesso incerto. Fra queste proteine si possono ricordare la β- e la γ-trace, l’α1-microglobulina, l’α2-microglobulina (o proteina legante il retinolo), e la β2-microglobulina.
L’α1-microglobulina è una proteina di 31 kDa di origine epatica la cui concentrazione nel plasma si riduce soprattutto nelle cirrosi scompensate e nell’epatite fulminante, mentre aumenta in alcune neoplasie come il mieloma.
L’α2-microglobulina forma nel plasma un complesso di 85 kDa con la vitamina A e la prealbumina (vedi Par. 8.1.2). Poiché è eliminata per via renale, la sua concentrazione plasmatica aumenta con la riduzione del filtrato glomerulare. Questa proteina diminuisce nel plasma nel corso dei processi infiammatori e in caso di deficit di vitamina A.
La β2-microglobulina (11 kDa) costituisce la catena leggera dell’antigene maggiore di istocompatibilità di classe I presente sulla superficie di tutte le cellule nucleate da cui viene liberata nel plasma assieme ad un’altra frazione secreta dalle cellule linfoidi. Per il basso peso molecolare non viene trattenuta dal filtro glomerulare. Tuttavia, viene in gran parte riassorbita e successivamente metabolizzata a livello del tubulo renale, per cui solo lo 0,1% della quantità ultrafiltrata viene escreta con le urine. I valori di riferimento nel siero sono compresi fra 1-2 mg/L; la vita media è inferiore ai 100 minuti. Un aumento nel siero della β2-microglobulina può essere messo in relazione con una diminuita capacità di filtrazione glomerulare. Tuttavia, questa proteina aumenta nel siero anche in corso di malattie sistemiche autoimmuni, AIDS, gammapatie monoclonali maligne, o carcinomi (del tubo gastroenterico, mammella, polmone, ovaio, utero, etc.).
8.5. COMPLEMENTO
Il complemento costituisce un importante mediatore nei processi infiammatori e di difesa dell’organismo. Esso è formato da un insieme di proteine sintetizzate negli epatociti o in vari distretti del sistema macrofagico, caratterizzate da una breve emivita e assommanti nella loro globalità a circa il 10% delle globuline del siero umano normale, appartenendo per lo più alle frazioni β1 e β2. La maggior parte di queste proteine esiste nel siero umano sotto forma di un precursore inattivo.
8.5.1. Funzione biologica
Le proteine del complemento sono attivate in modo sequenziale (attraverso una via classica o una via alternativa), formando prodotti intermedi che esplicano diverse azioni biologiche. Inoltre, le componenti terminali della cascata complementare hanno la capacità di determinare lesioni irreversibili sulle membrane cellulari, contribuendo da un lato alla difesa contro i microorganismi e dall’altro lato alla patogenesi dei processi morbosi derivanti da una abnorme risposta del sistema immunitario.
8.5.1a. La via classica di attivazione
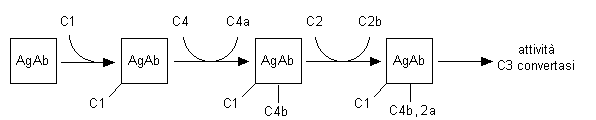
Fig. 8.1. Via classica di attivazione del complemento
La via classica è generalmente attivata da quegli
immunocomplessi in cui intervengono le immunoglobuline IgG1, IgG2,
IgG3 e IgM (vedi Par. 8.6.1). Tuttavia anche la proteina C reattiva e alcuni RNA-virus,
micoplasmi e batteri (sia gram-positivi che gram-negativi)
possono attivare il complemento in questo modo. L’attivazione
è promossa dal legame dei fattori sopraindicati alla frazione C1
del complemento (Fig. 8.1). Questa frazione
è costituita da tre subcomponenti C1q, C1r e C1s nel rapporto
molare di 1:2:2. La subcomponente C1q (a sua volta formata da 18
subunità: 6 catene A, 6 catene B e 6 catene C) lega il fattore
attivante inducendo, mediante un processo autocatalitico, la
scissione della componente C1r in un eterodimero legato da un
ponte disulfuro (C1r attivato) ![]() . Il C1r attivato si comporta come una
endopeptidasi capace di scindire la componente C1s in due
frammenti. Questi rimangono legati da un ponte disulfuro e
costituiscono un prodotto di attivazione (C1s attivato) capace di
agire come una endopeptidasi sulla subunità a
della componente C4 e sulla componente C2.
. Il C1r attivato si comporta come una
endopeptidasi capace di scindire la componente C1s in due
frammenti. Questi rimangono legati da un ponte disulfuro e
costituiscono un prodotto di attivazione (C1s attivato) capace di
agire come una endopeptidasi sulla subunità a
della componente C4 e sulla componente C2.
La componente C4 nativa è a sua volta formata da tre subunità (α, β e γ). A seguito dell’azione del C1s attivato, la componente C4 si scinde in un frammento più piccolo (C4a) e in uno più grande (C4b) che espone sulla superfice del residuo della subunità a un legame tioestere altamente labile, destinato ad idrolizzarsi rapidamente o a reagire, attraverso il suo gruppo acile, con un gruppo ossidrile di un polisaccaride o con un gruppo aminico di una proteina, ancorando covalentemente la componente C4b alla superficie cellulare o alle immunoglobuline (Fig. 8.2). La componente C2 nativa è più efficientemente scissa dal C1s attivato dopo essersi legata alla componente C4b in quanto questa si trova già in prossimità del C1s attivato sull’immunocomplesso. Come conseguenza dell’azione endopeptidasica esercitata dal C1s attivato sul C2, si ha la liberazione del frammento C2b e la formazione del complesso attivato C4b,2a con attività C3 convertasi, capace cioè di agire come una endopeptidasi sulla subunità α della componente C3 (un dimero formato da una catena α e una β), scindendolo nei frammenti C3a e C3b.
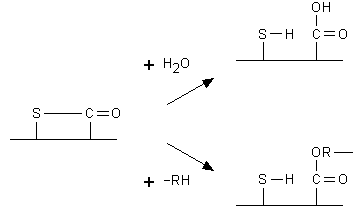
Fig. 8.2. Scissione del legame tioestere nelle componenti C4b e C3b. La presenza di un gruppo ossidrile o aminico (-RH) sul complesso antigene-anticorpo permette l’ancoraggio covalente della componente del complemento.
8.5.1b. La via alternativa di attivazione
La via alternativa non richiede la presenza di immunocomplessi per la sua attivazione e può perciò essere vista come un importante meccanismo di immunità naturale. La componente C3 va incontro infatti ad un lento processo di idrolisi spontanea di un legame tioestere intramolecolare che la trasforma in una molecola "C3b-simile" (Fig. 8.3). Questa è capace di legare il fattore B e renderlo suscettibile all’azione endopeptidasica esercitata dal fattore D (una endopeptidasi prodotta dai monociti e dai macrofagi probabilmente già in forma attiva) che lo scinde nei frammenti Ba e Bb. Il complesso C3,Bb attivato, chiamato anche C3 convertasi "da innesco", è responsabile di una lenta e continua scissione della componente C3 nei frammenti C3a e C3b. Il frammento C3b nascente contiene, al pari del frammento C4b precedentemente descritto (Fig. 8.2), un legame tioestere particolarmente reattivo che può andare incontro a un processo di transesterificazione o di amidazione con gruppi presenti sulla superficie cellulare o, quando questi sono assenti, ad una idrolisi spontanea (con susseguente inattivazione del C3b stesso). Il frammento C3b legato ad una opportuna superficie forma a sua volta un complesso con il fattore B nativo, rendendolo substrato del fattore D e formando con il suo prodotto di scissione un sistema enzimatico particolarmente efficiente, C3b,Bb attivato, chiamato C3-convertasi "amplificante" (capace anch’esso di scindere il C3 in C3a e C3b). Si viene a creare così un sistema ciclico di amplificazione della risposta in quanto il C3b è nello stesso tempo parte della C3-convertasi e prodotto finale della via alternativa. Questo sistema può attivarsi autonomamente o essere usato per incrementare il segnale della via classica di attivazione del complemento.
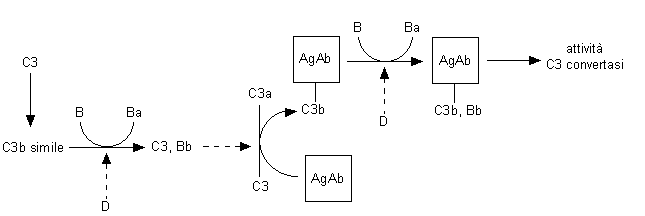
Fig. 8.3. Via alternativa di attivazione del complemento
8.5.1c. Il complesso terminale litico
Come abbiamo visto, entrambe le vie di attivazione del complemento portano alla formazione di convertasi capaci di agire sulla componente C3. Il legame del C3b alle C3-convertasi della via classica o alternativa porta rispettivamente alla formazione dei complessi C4b,2a,3b attivato e (C3b)2,Bb attivato e rende queste convertasi specifiche per la componente C5 (un dimero formato da una catena α e una β).
L’attivazione della componente C5, dovuta all’azione endopeptidasica di una delle due C5-convertasi sulla sua subunità a, porta alla formazione del prodotto a maggiore peso molecolare C5b e alla liberazione del frammento C5a. Se il C5b si lega alla componente C6 prima ancora di staccarsi dalla C5-convertasi, esso viene stabilizzato nella sua sede di ancoraggio e reso capace di legarsi in successione alle componenti C7, C8 e C9 (che a sua volta polimerizza). Si viene così a creare una macromolecola che inserendosi nella membrana cellulare forma in essa un canale a tutto spessore che causa la lisi della cellula.
8.5.1d. I prodotti intermedi di attivazione
Due tipi di prodotti intermedi si formano nel corso del processo di attivazione del complemento: i frammenti diffusibili a basso peso molecolare (C3a, C4a e C5a) e i frammenti ad alto peso molecolare (in particolare il C3b) legati al materiale da inattivare.
Recettori per i frammenti C3a, C4a e C5a sono presenti in numerose cellule. I frammenti C3a e C5a si comportano come anafilatossine (promuovono il rilascio di istamina dai basofili e dalle mast-cellule, l’attivazione e l’aggregazione dei neutrofili - con produzione di metaboliti dell’acido arachidonico e di radicali dell’ossigeno e con il rilascio degli enzimi dei granuli -, l’aggregazione delle piastrine e il rilascio da esse di metaboliti dell’acido arachidonico, la contrazione della muscolatura liscia e l’aumento della permeabilità vasale). Il frammento C5a è inoltre un potente fattore chemiotattico per i leucociti polimorfonucleati e i monociti. Sia il C3a che il C5a sono in gran parte inattivati da una carbossipeptidasi presente nel siero che scinde una arginina C-terminale presente in entrambe le molecole.
I recettori per il frammento C3b e per i suoi prodotti di idrolisi sono di tre tipi (CR1, CR2 e CR3) e sono presenti in diversa misura sugli eritrociti, leucociti polimorfonucleati, monociti, linfociti, macrofagi, mast-cellule e sui podociti glomerulari (vedi Par. 11). Il frammento C3b si comporta come una potente opsonina capace di promuovere la fagocitosi dei microorganismi. Esso ostacola inoltre la precipitazione degli immunocomplessi, promuovendone nel contempo la solubilizzazione e la rimozione sia attraverso l’attivazione dei macrofagi sia legando questi complessi agli eritrociti circolanti.
8.5.1e. Le proteine di controllo
Le vie di attivazione classica e alternativa sono sotto il controllo di diversi sistemi modulatori. L’azione della componente C1 è inibita da una proteina (C1-INH) capace di legarsi covalentemente alle subcomponenti C1r e C1s provocandone la dissociazione dal complesso macromolecolare. La proteina legante il C4 (C4-bp; C4-binding protein) e il fattore accelerante il decadimento (DAF; decay-accelerating factor) inibiscono il C4b,2a attivato accelerando il distacco della componente C2a. Il fattore I inattiva il C4b e il C3b legati alle membrane rispettivamente mediante il distacco proteolitico dei frammenti C4d e iC3b. L’azione inibente del fattore I è notevolmente accellerata dal C4-bp e dal fattore H. Il fattore H infine compete con il fattore B nel legame con il C3b ed è inoltre capace di dissociare il complesso attivato C3b,Bb.
Sono noti diversi attivatori della via alternativa del complemento. La properdina agisce in tal senso impedendo la dissociazione del complesso C3b,Bb attivato. Le cellule di lievito, gli eritrociti di montone ed alcuni batteri si legano al C3b nascente e proteggono il C3b,Bp attivato dall’inattivazione dovuta ai fattori H ed I. Anche gli anticorpi, pur non essendo necessari all’attivazione della via alternativa, possono partecipare funzionalmente a questo processo attraverso il legame con batteri o cellule infettate da virus mediante meccanismi diversi da caso a caso.
8.5.2. Variazioni dell’attività complementare
Il dosaggio dell’attività complementare totale o delle singole componenti può essere utile per individuare specifiche carenze congenite o un’eventuale attivazione della via classica o alternativa del sistema.
Per quasi tutti i fattori sono stati descritti rari casi di difetti ereditari (Tab. 8.I). Questi generalmente si trasmettono per via autosomica recessiva ad eccezione del deficit di C1-INH (autosomico dominante) e di properdina (legato al cromosoma X).
Tab. 8.I. Deficit ereditari dei fattori del complemento.
| Deficit | Trasmissione ereditaria |
Manifestazioni cliniche |
| C1q | autosomica recessiva |
lupus
|
| C1r/s | autosomica recessiva |
lupus, glomerulonefrite |
| C2 | autosomica recessiva |
lupus, infezioni da piogeni |
| C3 | autosomica recessiva |
lupus, infezioni da piogeni |
| C4A / C4B | autosomica recessiva |
lupus, infezioni da piogeni |
| C5 | autosomica recessiva |
sepsi meningococcica, meningite |
| C6 | autosomica recessiva |
sepsi meningococcica, meningite |
| C7 | autosomica recessiva |
sepsi meningococcica, meningite |
| C8 | autosomica recessiva |
sepsi meningococcica, meningite |
| C9 | autosomica recessiva |
asintomatico |
| C1-IHN | autosomica dominante |
angioedema |
| C4-bp | sconosciuta |
angioedema |
| DAF | sconosciuta |
fenotipo inab |
| Fattore D | sconosciuta |
sepsi meningococcica, meningite, polmonite |
| Fattore H | autosomica recessiva |
sindrome emolitica uremica |
| Fattore I | autosomica recessiva |
infezioni da piogeni |
| Properdina | legata al cromosoma X |
sepsi meningococcica, meningite |
8.6. LE IMMUNOGLOBULINE
Le immunoglobuline (Ig) sono delle glicoproteine costituite,
nella loro forma monomerica, dall’unione di quattro catene
polipeptidiche uguali a due a due, unite da ponti disulfuro e di
diversa massa molecolare: due catene pesanti (H) di 50 kDa e due
catene leggere (L) di 25 kDa. Ciascuna catena è costituita da
una regione variabile (VH e VL) e da una
regione costante (CH e CL). Le regioni
variabili, comprendenti il tratto N-terminale delle catene per
una lunghezza di 100-200 residui, concorrono alla formazione dei
siti combinatori a cui si lega l’antigene specifico e sono
diverse per ogni singolo anticorpo (eterogeneità idiotipica). Le
regioni costanti, comprendenti le porzioni delle catene fra la
fine della regione variabile e l’estremo C-terminale, sono
caratterizzate da tratti di circa 100 residui aminoacidici con
sequenza omologa (domains): uno nella catena L (CL)
e tre nella catena H (CH1, CH2 e CH3).
I tratti CH1 e CH2 delle catene pesanti
sono uniti da una segmento di 10-15 aminoacidi (hinge region)
contenente le cisteine che partecipano ai legami disolfuro
intercatena e, nelle IgG, un elevato numero di proline
![]() .
.
8.6.1. Eterogeneità isotipica
Le immunoglobuline possono essere divise in isotipi sulla base
di differenze fisico-chimiche ed antigeniche nella regione
costante delle catene pesanti e leggere. Le differenti catene
pesanti (γ1, γ2,
g3, γ4, μ, α1, α2, δ ed ε) caratterizzano le classi e le sottoclassi
delle immunoglobuline: IgG1, IgG2, IgG3, IgG4, IgM, IgA1, IgA2,
IgD e IgE
![]() . Le
differenti catene leggere (κ e λ) permettono di distinguere ciascuna classe
o sottoclasse in due tipi (non è possibile infatti che le catene κ e λ siano
presenti contemporaneamente nella stessa immunoglobulina).
. Le
differenti catene leggere (κ e λ) permettono di distinguere ciascuna classe
o sottoclasse in due tipi (non è possibile infatti che le catene κ e λ siano
presenti contemporaneamente nella stessa immunoglobulina).
Normalmente sono sintetizzate più catene κ che λ (il rapporto κ/λ è uguale a 1,2-2,0) e più catene leggere che pesanti in modo tale che una quota di catene leggere policlonali libere è sempre presente nel siero.
8.6.1a. Immunoglobuline G
Le IgG nel loro insieme rappresentano il 70-80% delle immunoglobuline del siero. Il loro aumento a seguito di una stimolazione antigenica primaria segue quello delle IgM. Però, una successiva stimolazione antigenica determina un prevalente incremento di IgG. Le IgG sono praticamente le uniche immunoglobuline che passano dalla madre al feto attraverso la placenta. Le IgG materne costituiscono così la maggior quota delle immunoglobuline presenti nel siero del neonato fino a quando la produzione endogena di IgG diviene efficace verso il 4°-6° mese di vita.
La numerazione delle sottoclassi rispecchia la loro concentrazione relativa: IgG1, 60-70% ; IgG2, 15-20%; IgG3, 5-8%; IgG4, 1-5%. L’emivita è grossolanamente direttamente proporzionale alla concentrazione e, in media, è di 3-4 settimane. Le differenze nella sequenza aminoacidica fra le diverse IgG sono particolarmente evidenti a livello della hinge region, più lunga nelle IgG3. Tutte le quattro sottoclassi partecipano alla risposta immunitaria; tuttavia, quando l’antigene è di natura proteica, vengono prodotte principalmente delle IgG1 e IgG3, mentre le IgG2 sono più abbondanti nella risposta verso antigeni che contengono carboidrati. Le IgG1, IgG2 e IgG3 si legano al complemento a livello del domain CH2, attivando la via classica, mentre le IgG4 contribuiscono ad innescare la via alternativa. Le IgG1 e le IgG3 promuovono con maggiore efficienza l’opsonizzazione microbica in quanto hanno maggiore affinità rispetto le altre sottoclassi per i recettori dei macrofagi e dei neutrofili a cui si legano attraverso il domain CH3 promuovendo la chemiotassi, la fagocitosi e il rilascio di mediatori citotossici.
8.6.1b. Immunoglobuline M
Le IgM si trovano generalmente nel siero sotto forma di un pentamero di 900 kDa a simmetria radiale stabilizzato da legami disolfuro ed unito ad un polipeptide J di 15 kDa che funge da ponte fra le diverse unità monomeriche. Tuttavia le IgM possono trovarsi anche in forma monomerica nel siero in varie condizioni di ipergammaglobulinemia e sulla superficie dei linfociti B. Le IgM sono i primi anticorpi che si formano durante la vita fetale, ma non sono capaci di attraversare la barriera placentare: un loro aumento nel neonato è perciò indice di un’infezione congenita. Nell’adulto le IgM rappresentano l’8-10% del totale delle immunoglobuline. Sono le prime ad aumentare nella risposta primaria alla somministrazione di un antigene, ma a causa della breve emivita (5 giorni) non contribuiscono molto al mantenimento dell’immunità in assenza di un’esposizione cronica all’antigene. Valori di IgM particolarmente elevati vengono raggiunti con l’esposizione ad antigeni polisaccaridici (endotossine dei batteri Gram-negativi, antigeni eritrocitari del sistema AB0, etc.). Le IgM pentameriche sono più efficienti rispetto le immunoglobuline monomeriche nel promuovere l’opsonizzazione e l’agglutinazione batterica così come l’attivazione della via classica del complemento. Una piccola quota di IgM è presente nelle secrezioni esocrine, dove aumenta nel caso di deficit selettivo di IgA supplendo così alla mancanza di questa classe di immunoglobuline.
8.6.1c. Immunoglobuline A
Le IgA (IgA1 e IgA2) costituiscono il 7-17% delle immunoglobuline nel siero, dove hanno un’emivita di 6 giorni, ma rappresentano la classe prevalente di immunoglobuline nelle secrezioni (saliva, latte, secreto bronchiale ed intestinale). Le IgA sieriche sono prevalentemente dei monomeri, mentre le IgA delle secrezioni sono in massima parte dei dimeri legati al polipeptide J (vedi Par. 8.6.1b) e ad un altro polipeptide, denominato secretory component, di 60 kDa che sembra avere un ruolo nel facilitare il trasporto delle IgA sul versante esterno delle cellule epiteliali, una volta prodotte dalle plasmacellule presenti nella lamina propria. Le IgA sono raramente prodotte dal feto in assenza di infezioni congenite e raggiungono per ultime (verso i 12-14 anni) i livelli propri dell’adulto. Le IgA non attivano la via classica del complemento, ma possono svolgere una funzione antibatterica nei secreti interagendo con componenti della via alternativa. Le IgA svolgono un compito importante nella risposta immunitaria a vaccini somministrati per via orale o nasale, nell’impedire l’entrata di antigeni attraverso le mucose e nel rimuovere gli antigeni presenti in circolo attraverso il sistema biliare.
8.6.1d. Immunoglobuline D
Le IgD sono presenti nel siero in bassa concentrazione (0,1-0,2% delle immunoglobuline totali). Esse si trovano, insieme alle IgM, sulla membrana della maggior parte dei linfociti B ove sembra che rappresentino il recettore per il primo contatto con l’antigene. Questo riconoscimento darebbe luogo ad un segnale per la successiva proliferazione del clone B e la sua differenzazione in plasmacellule.
8.6.1e. Immunoglobuline E
Le IgE sono le immunoglobuline meno concentrate nel plasma ed hanno un’emivita di 2-3 giorni. Non attraversano la barriera placentare, ma possono essere sintetizzate in modo limitato dal feto; raggiungono il livello proprio degli adulti attorno ai 10 anni. Le IgE si fissano a recettori specifici presenti sulla membrana dei basofili e dei mastociti. Quando i siti combinatori delle IgE vengono a contatto con l’antigene specifico, si innesca una serie di reazioni che portano alla liberazione dalle cellule di istamina e di altri modulatori dell’ipersensibilità. Le IgE aumentano nel siero in diverse situazioni patologiche e in particolare modo nei soggetti affetti da ipersensibilità a diversi allergeni o da parassitosi intestinale.
8.6.2. Variazioni dei livelli sierici delle immunoglobuline
I livelli sierici delle immunoglobuline presentano una notevole variabilità in relazione a fattori di ordine individuale o ambientale.
Tra i fattori individuali, particolare importanza ha l’età. La maggior parte delle immunoglobuline presenti nel neonato sono della classe IgG e di origine materna. Queste vengono sostituite dalle immunoglobuline neosintetizzate verso il 5° mese. Al primo anno di vita, le immunoglobuline sono circa il 50% rispetto all’adulto, i cui valori vengono raggiunti solo dopo i 16 anni. Nell’anziano la concentrazione delle immunoglobuline è significativamente più elevata rispetto all’adulto.
Per quanto riguarda i fattori ambientali, è da ricordare che la risposta immunitaria è influenzata dalla frequenza, dall’intensità e dal tipo di stimolazione antigenica: la risposta primaria coinvolge le IgM, la secondaria le IgG; gli antigeni batterici incrementano le IgG, quelli virali le IgM e le IgA, quelli parassitari e pollinici le IgE.
Variazioni patologiche della concentrazione delle immunoglobuline sieriche possono verificarsi per una alterazione della sintesi o del catabolismo ed interessare tutte ovvero una singola classe immunoglobulinica.
8.6.2a. Immunodeficienze
Valori di IgG inferiori a 2 g/L e di IgM e IgA inferiori a 250 mg/L in presenza di una normale quota di albumina possono essere considerati indicativi di una condizione di immunodeficienza in soggetti di età superiore a 6 anni. Per quanto riguarda i bambini di età inferiore a 6 anni, devono essere considerati significativi solo i livelli di IgG inferiori a 1 g/L e di IgM e IgA inferiori a 50-100 mg/L.
Una ridotta sintesi dell’immunoglobuline può dar luogo a deficit immunologici globali con quadri dominati da processi infettivi particolarmente gravi e spesso precocemente letali: nell’immunodeficienza grave combinata, dovuta a un difetto della cellula staminale, e nell’agammaglobulinemia associata a timoma l’assenza di Ig circolanti si accompagna ad un deficit anche della linea cellulare T; nell’agammaglobulinemia congenita legata al cromosoma X il difetto interessa il differenziamento delle cellule pre-B in B e pertanto il numero e la funzione dei linfociti T rimangono inalterati; nelle ipogammaglobulinemie secondarie (a neoplasie, a infezioni, a trattamento con immunosoppressori, etc.) vi è un’incapacità delle cellule B a proliferare e a trasformarsi in plasmacellule produttrici di Ig.
Una ridotta sintesi interessante singole classi
immunoglobuliniche si osserva nel caso di deficit selettivo di
IgM, IgA, IgE o delle varie sottoclassi delle IgG e nella
sindrome di Wiskott-Aldrich ![]() . Una ridotta sintesi con interessamento
contemporaneo di più classi immunoglobuliniche si ha nella disgammaglobulinemia tipo I
(con deficit di IgG
ed IgA) e nell’atassia telangectasica
. Una ridotta sintesi con interessamento
contemporaneo di più classi immunoglobuliniche si ha nella disgammaglobulinemia tipo I
(con deficit di IgG
ed IgA) e nell’atassia telangectasica
![]() (spesso con deficit di
IgA ed IgE).
(spesso con deficit di
IgA ed IgE).
Deficit immunoglobulinici dovuti ad eccessiva perdita di Ig
sono osservabili nella sindrome nefrosica
![]() (dove diminuiscono
prevalentemente le IgG e le IgA) e nelle gastroenteropatie
proteinodisperdenti (dove la perdita è meno selettiva). In altri
casi, il deficit può derivare da un accelerato catabolismo
proteico come nell’ipoproteinemia familiare, nella distrofia miotonica
(dove diminuiscono
prevalentemente le IgG e le IgA) e nelle gastroenteropatie
proteinodisperdenti (dove la perdita è meno selettiva). In altri
casi, il deficit può derivare da un accelerato catabolismo
proteico come nell’ipoproteinemia familiare, nella distrofia miotonica
![]() o in seguito a terapia con
gammaglobuline in soggetti con difetti selettivi per formazione
di anticorpi specifici verso la classe immunoglobulinica carente.
o in seguito a terapia con
gammaglobuline in soggetti con difetti selettivi per formazione
di anticorpi specifici verso la classe immunoglobulinica carente.
8.6.2b. Iperimmunoglobulinemie
Una alterazione delle immunoglobuline per eccesso si verifica in diverse situazioni che comportano un aumento delle gammaglobuline, come ad esempio nelle infezioni croniche, nelle malattie autoimmuni, nelle epatopatie croniche e negli stadi avanzati dei tumori maligni. In questi casi l’aumento di produzione delle immunoglobuline interessa contemporaneamente numerosissimi stipiti cellulari immunocompetenti, cosicché le immunoglobuline sono interessate nel loro complesso, anche se a carico delle IgG si verificano le modificazioni più marcate (gammopatie policlonali).
Un altro gruppo di malattie è caratterizzato da un fortissimo incremento di gammaglobuline che non solo è circoscritto ad una sola classe immunoglobulinica, ma che presenta anche caratteri così elevati di omogeneità da permettere di correlare l’alterazione del quadro plasmatico alla proliferazione di un solo stipite di cellule immunopoietiche (gammopatie monoclonali). Le gammopatie monoclonali sono nella maggior parte dei casi benigne ed hanno una incidenza inferiore all’1% (nei soggetti con meno di 25 anni) o attorno al 4% (nei pazienti con più di 70%). Nel rimanente 25% dei casi, tuttavia, le gammopatie monoclonali sono espressione di un grave disordine linfoproliferativo in atto o in progressiva evoluzione (mieloma multiplo, malattia delle catene pesanti, leucemia linfatica cronica, macroglobulinemia di Waldenström).
Le discrasie del sistema immunitario possono essere caratterizzate dalla presenza di crioglobuline in circolo. Queste sono classificate in tre tipi. Le crioglobuline tipo I sono costituite da immunoglobuline che precipitano a freddo per le loro peculiari caratteristiche fisico-chimiche. Le crioglobuline tipo II e III sono immunocomplessi costituiti da un antigene rappresentato da immunoglobuline per lo più di classe IgG e da un fattore reumatode monoclonale (nel tipo II) o policlonale (nel tipo III) rappresentato dalle IgM. Le crioglobulinemie di tipo I e II sono associate a malattie linfoproliferative e autoimmuni, mentre le crioglobulinemie di tipo III sono presenti in una grande varietà di malattie infettive, epatiche ed autoimmuni. Fra le crioglobulinemie di tipo III è da ricordare la crioglobulinemia mista essenziale (sindrome di Lospalluto-Meltzer), particolarmente frequente nell’Europa meridionale ed in Italia e caratterizzata da porpora con localizzazione elettiva agli arti inferiori e da una vasculite sistemica che interessa arterie e vene di piccolo calibro. Nel 70-91% dei soggetti con crioglobulinemia mista essenziale sono evidenziabili segni di esposizione al virus dell’epatite C.
8.7. VALUTAZIONE DEL QUADRO PROTEICO
La determinazione della velocità di eritrosedimentazione costituisce un metodo semplice, poco costoso e di frequente uso per valutare grossolane variazioni della composizione proteica del plasma, quali sono quelle che possono comparire nelle reazioni di fase acuta, nelle malattie infiammatorie acute e croniche o nelle neoplasie, specie in presenza di metastasi epatiche. Questo esame consiste nel misurare la velocità con cui la parte corpuscolata del sangue sedimenta separandosi dalla parte liquida, quando il sangue, opportunamente diluito con un anticoagulante, viene collocato in una provetta lunga e stretta. La velocità di eritrosedimentazione dipende dalle dimensioni degli elementi corpuscolati, dalla loro densità relativa rispetto al plasma e dalla viscosità di quest’ultimo. Il principale meccanismo che porta ad un aumento della velocità di eritrosedimentazione è la tendenza dei globuli rossi ad impilarsi fra loro, formando dei voluminosi aggregati (rouleaux). La formazione dei rouleaux è condizionata in gran parte dalle caratteristiche fisico-chimiche del plasma e, in particolare, dalla sua composizione proteica. La formazione dei rouleaux e, conseguentemente, l’incremento della velocità di eritrosedimentazione sono favoriti quando aumentano nel plasma le proteine che hanno una struttura asimmetrica, come ad esempio il fibrinogeno, le α-globuline e le γ-globuline. La velocità di eritrosedimentazione tende invece a diminuire quando sono presenti delle alterazioni morfologiche sulle cellule (anisocitosi, sferocitosi; vedi Par. 7.1.3) o quando è aumentata la viscosità del sangue. La velocità di eritrosedimentqazione è notevolmente aumentata nelle anemie gravi, mentre è ritardata nelle policitemie.
L’analisi elettroforetica delle sieroproteine è un altro degli esami più frequentemente richiesti. Nell’età infantile è utile, a scopo di orientamento, soprattutto per la individuazione di eventuali anomalie genetiche e delle complicanze concomitanti o per valutare la risposta delle proteine della fase acuta. Nell’età matura ed avanzata, l’analisi viene richiesta soprattutto per l’individuazione di eventuali gammopatie policlonali o monoclonali acquisite e di eventuali ipogammaglobulinemie.
Le variazioni abnormi o patologiche delle bande proteiche separate elettroforeticamente vengono valutate sia sotto un aspetto qualitativo che quantitativo. La lettura fotodensitometrica delle bande, opportunamente colorate, consente infatti di stabilire il loro diverso rapporto percentuale. A ciascuna frazione viene quindi attribuita una quota ponderale espressa in g/L sulla base del valore della proteinemia totale, che è determinato a parte e, di regola, con metodi chimici. Queste operazioni di calcolo, semiquantitative o addirittura arbitrarie, sebbene siano entrate nell’uso, sono soggette a sostanziali obiezioni in quanto (1) non tutte le bande sono monoproteiche, (2) il colorante ha una diversa affinità per le diverse proteine, (3) i dosaggi immunochimici danno valori alquanto diversi da quelli desumibili dall’esame elettroforetico (soltanto per l’albumina e l’α1-antitripsina si ottengono valori abbastanza simili). Le discordanze analitiche sono da attribuirsi anche alla dispersione elettroforetica delle immunoglobuline, per cui i valori delle frazioni α2 e β risulterebbero elettroforeticamente superiori alla somma delle componenti plasmatiche interessate, mentre i valori delle gamma risulterebbero inferiori a quelli delle Ig totali determinate con metodi immunochimici.
Per questi motivi, nel 5-10% dei casi l’analisi elettroforetica è affiancata da ulteriori indagini, da eseguirsi talvolta oltre che sul siero anche sulle urine, che possono essere una elettroforesi ad alta risoluzione (come controllo qualitativo del reperto ad un livello molecolare più accurato), un dosaggio biochimico o immunochimico delle singole proteine, una tipizzazione delle bande monoclonali o oligoclonali mediante immunoelettroforesi e/o immunofissazione.
8.7.1. Metodi di determinazione
Il principio generale su cui si basa gran parte delle tecniche per il riconoscimento e la determinazione quantitativa delle singole frazioni proteiche è costituito dalla reazione antigene-anticorpo che si esegue cimentando il liquido biologico, in cui è contenuta la proteina in esame che funge d’antigene, con il relativo anticorpo. La reazione può avvenire in un mezzo liquido o semisolido (gel di agar o agarosio) ed è evidenziata con tecniche immunologiche ed immunochimiche.
Per quanto riguarda le proteine totali e l’albumina è possibile usare anche metodiche biochimiche specifiche che non prevedono l’utilizzo di anticorpi specifici.
8.7.1a. Elettroforesi delle proteine plasmatiche
L’elettroforesi delle proteine plasmatiche può essere effettuata utilizzando diversi tipi di supporto. Alcuni di questi (carta, acetato di cellulosa, agarosio) permettono la separazione delle proteine unicamente in base alla loro carica netta. Altri supporti (amido, poliacrilamide) formano un gel con pori paragonabili alle dimensioni delle proteine da separare, in modo che queste ultime possano essere discriminate sia in base alla carica netta che al peso molecolare. L’acetato di cellulosa ha numerosi vantaggi rispetto la carta (ormai raramente usata): è sufficiente un campione di siero molto piccolo, le diverse bande proteiche sono separate in maniera netta e in poco tempo, il supporto può essere reso trasparente dopo la colorazione permettendo una lettura densitometrica del colore. Il gel di agarosio offre i medesimi vantaggi dell’acetato di cellulosa, separando ancora più nettamente le bande, ma a causa della sua carica debolmente negativa in ambiente alcalino dà luogo ad un flusso elettro-osmotico che porta a spostare leggermente verso il catodo le proteine (γ-globuline) con pI vicino al pH del tampone. La poliacrilamide permette una separazione molto più dettagliata rispetto alle tecniche precedenti riuscendo a far individuare fino a 100 bande proteiche (contro le 5-13 bande osservabili in acetato di cellulosa o in agarosio), tuttavia il quadro elettroforetico appare spesso troppo complesso e di difficile interpretazione.
Il tampone comunemente usato nell’elettroforesi del siero è il barbital a pH 8,6. Altri tamponi proposti sono il Tris-barbital con lattato di calcio e il Tris-borato con EDTA. Il borato aggiunto al tampone interagisce con le glicoproteine migliorando la risoluzione dell’analisi. Questi tamponi conferiscono a gran parte delle proteine plasmatiche una carica netta negativa che le fa migrare verso l’anodo.
Le varie bande proteiche separate elettroforeticamente sono evidenziate con soluzioni coloranti contenenti sostanze (acido tricloroacetico, metanolo) capaci di fissare le proteine sul supporto. I coloranti più usati sono l’Amido Schwarz 10B, il Rosso Ponceau S, il Bromofenolo, il Coomassie Brilliant Blue R250. Quest’ultimo colora intensamente le bande proteiche ed è particolarmente adatto nel caso dell’elettroforesi delle proteine urinarie o del fluido cerebrospinale. In generale, i coloranti hanno maggiore affinità per l’albumina che per le globuline.
Una altra tecnica elettroforetica è l’elettrofocalizzazione delle proteine al punto isoletettrico su un gradiente di pH da 4 ad 8 su gel. Questa procedura è particolarmente usata per confermare la presenza di paraproteine nel siero o di bande monoclonali nel fluido cerebrospinale.
8.7.1b. Proteine totali
Il procedura di Kjeldahl è il più vecchio metodo per la determinazione dell’azoto proteico. Si basa sull’ossidazione delle materia organica precipitata in presenza di H2O2, H2SO4, K2SO4 (per elevare il punto di ebollizione dell’H2SO4) e CuSO4 (usato come catalizzatore) alla temperatura di 340-360°C in modo da trasformare tutto l’azoto proteico in solfato d’ammonio. Dopo aver aggiunto NaOH, l’ammoniaca viene fatta distillare in acido borico e titolata con HCl. La procedura è lenta, laboriosa e poco sensibile, pur rimanendo il più preciso ed accurato metodo per la determinazione dell’azoto proteico.
Il metodo del biureto sfrutta le proprietà cromoforiche del
complesso che si forma in alcali fra gli ioni Cu2+ e i
legami peptidici. Gli aminoacidi e i dipeptidi non danno questa
reazione, mentre tutti i peptidi più lunghi vengono determinati
![]() . Il metodo è semplice
e attendibile. Possibili interferenze si hanno con sieri itterici
o lipemici o quando il paziente è stato trattato con infusioni
di destrano.
. Il metodo è semplice
e attendibile. Possibili interferenze si hanno con sieri itterici
o lipemici o quando il paziente è stato trattato con infusioni
di destrano.
Il metodo di Lowry si basa sul fatto che i fenoli riducono il complesso fosfotungstomolibdico (reagente di Folin-Ciocalteu) in un prodotto di intenso colore blu. Nelle proteine la reazione è essenzialmente dovuta alla presenza di residui di tirosina. Le cisteine, le cistine e le istidine sono anch’esse reattive ma in misura molto minore. Il metodo è centinaia di volte più sensibile del metodo del biureto, ma manca in specificità. Inoltre, poiché il contenuto in tirosine varia nelle diverse proteine plasmatiche, anche l’intensità del colore prodotto può variare. Ad esempio le gammaglobuline sviluppano un colore più intenso del 23% rispetto l’albumina.
La concentrazione proteica può anche essere stimata sulla base dell’assorbimento del campione nell’ultravioletto, che è dovuto agli anelli aromatici delle fenilalanine, tirosine e triptofani nella regione compresa fra 270 nm e 290 nm e ai legami peptidici nella regione compresa fra 200 nm e 225 nm. Tuttavia, poiché il contenuto in aminoacidi aromatici varia nelle diverse proteine e poiché la composizione proteica varia nei diversi pazienti, la misura dell’assorbimento nella regione fra 270 nm e 290 nm non è adatta per gli scopi clinici. L’assorbimento nella regione fra 200 nm e 225 nm è molto più costante e circa 20 volte più elevato e permette di ottenere misure comparabili con quelle del metodo del biureto, qualora si abbia a disposizione uno spettrofotometro capace di operare nel lontano ultravioletto.
Un ulteriore metodo rapido e semplice per stimare la concentrazione delle proteine totali nel siero è quello di misurare la rifrazione della luce incidente. Lo strumento deve essere calibrato con sieri a titolo noto e deve essere usato a temperatura controllata. Il metodo porta tuttavia a stime errate nel caso di sieri lipemici, ipercolesterolemici, iperglicemici, iperbilirubinemici, iperazotemici, ipoproteinemici o emolizzati.
8.7.1c. Albumina
Le metodiche più comunemente usate per la determinazione dell’albumina si basano sulla proprietà che ha questa molecola di legare coloranti anionici. Poiché il colorante legato all’albumina ha uno spettro di assorbimento diverso da quello libero, è possibile effettuare la misura anche in presenza di un eccesso di reattivo. I coloranti più frequentemente usati sono il metilarancio, l’HABA (acido 2-(4’-idrossiazobenzene) benzoico), il verde di bromocresolo (3,3’,5’-tetrabromo-m-cresolsulfonftaleina, BCG) e il violetto di bromocresolo (5,5’-dibromo-o-cresolsulfonftaleina, BCP).
Il metodo al metilarancio non è specifico in quanto il colorante si lega anche alle β-lipoproteine e alle α1- e α2-globuline dando luogo ad una sovrastima dell’analita quando la sua concentrazione è bassa. Il metodo che utilizza l’HABA è più specifico per l’albumina; tuttavia ha una scarsa sensibilità e subisce l’interferenza di numerosi farmaci (salicilati, sulfamidici, penicillina, eparina). Il verde di bromocresolo ha una sufficiente sensibilità ma è ugualmente aspecifico, determinando una sovrastima dell’albumina specialmente in sieri con un anomalo rapporto fra le diverse frazioni proteiche (l’emoglobina, le α - e β-globuline e il fibrinogeno interferiscono nel saggio). Il violetto di bromocresolo è il più specifico fra i coloranti usati; la sovrastima dell’analita dovuta all’interferenza da eparina può essere eliminata aggiungendo dell’esadimetrina bromuro alla miscela di reazione.
Un altro colorante che si può adoperare per determinare l’albumina è il blu di bromofenolo. Il saggio viene eseguito a pH acido in modo che tutto il blu di bromofenolo in soluzione sia nella forma non ionizzata di colore giallo: se è presente della albumina, questa, legandosi esclusivamente alla forma ionizzata (blu) del colorante, sposta l’equilibrio verso questa specie ionica determinando il viraggio della miscela di reazione. Il saggio è generalmente eseguito su un supporto solido (Albustix) ed è particolarmente utile per la determinazione semiquantitativa di relativamente basse concentrazioni di albumina (ad esempio nelle urine). Risultati falsamente positivi possono essere imputati alla presenza di tamponi alcalini nel campione.
Molto meno usato è il metodo indiretto per la determinazione dell’albumina che si basa sul dosaggio quantitativo delle globuline, sfruttando il fatto che queste ultime hanno un contenuto in triptofano molto più elevato rispetto all’albumina. Il triptofano viene dosato fotometricamente in presenza di acido gliossilico e ioni calcio; la concentrazione dell’albumina è calcolata sottraendo la concentrazione delle globuline a quella delle proteine totali, determinate separatamente.
Altri metodi per la determinazione dell’albumina si basano sulla separazione elettroforetica delle diverse frazioni plasmatiche su acetato di cellulosa o agarosio. Dopo opportuna colorazione, la concentrazione relativa delle singole frazioni è determinata mediante un densitometro; la concentrazione dell’albumina è quindi calcolata moltiplicando la sua concentrazione relativa per la concentrazione totale delle proteine plasmatiche, misurata separatamente. La tecnica, oltre ad essere laboriosa e di difficile automazione, porta ad una sovrastima dell’analita in quanto i coloranti usati per evidenziare le varie bande elettroforetiche non legano le proteine in eguale misura, ma hanno generalmente una maggiore affinità per l’albumina.
I metodi immunologici per la determinazione dell’albumina si basano su tecniche di immunodiffusione radiale, di elettroimmunodiffusione, di radioimmunologia o di immunoenzimologia. L’immunocomplesso può essere inoltre dosato con un nefelometro. Quest’ultima tecnica è particolarmente adatta per essere applicata ad apparecchi automatici di analisi. Tutti i metodi immunologici hanno in comune lo svantaggio dell’alto costo dei reattivi per le analisi.
8.7.2. Preparazione del campione ed intervalli di riferimento
La velocità di eritrosedimentazione è determinata su sangue intero, diluito in un rapporto 4:1 con un anticoagulante (generalmente citrato di sodio). L’effettuazione dell’esame dopo 3 ore dal prelievo può determinare risultati non corretti. E’ consigliabile eseguire il prelievo con paziente a digiuno da almeno 8 ore.
Il siero è adoperato più comunemente rispetto al plasma nell’analisi qualitativa e quantitativa delle proteine. Il fibrinogeno contenuto nel plasma è infatti di difficile conservazione e dà luogo nell’elettroforesi ad una banda aggiuntiva (banda φ) che può rendere più difficoltosa l’interpretazione del tracciato. Bisogna inoltre ricordare che il contenuto in proteine del plasma è 2-4 g/L più alto rispetto al siero per la presenza del fibrinogeno.
La concentrazione delle proteine nel siero è compresa nel soggetto normale fra 66,6 g/L e 81,4 g/L. Per quanto riguarda le singole frazioni proteiche si rimanda a quanto detto nei paragrafi precedenti.
L’elettroforesi delle proteine consente di stimare simultaneamente almeno sette proteine che hanno nel siero normale maggiore rilevanza quantitativa. Queste proteine sono comunemente distribuite in cinque bande distinte. La banda A è praticamente monoproteica ed è costituita dall’albumina. La banda α1, anch’essa monoproteica, è costituita dalla α1-antitripsina. La banda α2 è costituita dalla α2-macroglobulina, generalmente nella porzione anodica, e dalla aptoglobina, generalmente nella porzione catodica. La banda β è costituita dalla transferrina, generalmente nella porzione anodica, e dalla componente C3 del complemento, generalmente nella porzione catodica. La banda γ è costituita dalle immunoglobuline delle diverse classi. E’ importante comunque ricordare che quote immunoglobuliniche migrano, anche nel normale, in posizioni anodiche del tracciato, comprese fra le zone α2, β e φ, posizione quest’ultima intermedia tra β e γ in cui migra il fibrinogeno (se è presente).
Metodiche a più alta risoluzione permettono la separazione delle componenti presenti nelle α2 e nelle β , nonché di evidenziare la prealbumina in posizione anodica rispetto l’albumina. Altre componenti del siero come le lipoproteine o l’emopessina, pur di una certa rilevanza quantitativa, non sembrano incidere sensibilmente sull’aspetto e l’intensità delle bande colorate con i comuni procedimenti (Amido Schwarz 10B, Rosso Ponceau S). Nella Tab. 8.II è riportato il rapporto fra le diverse frazioni del siero evidenziabili con una elettroforesi su gel di agarosio.
Tab. 8.II. Elettroforesi delle siero proteine. Rapporto percentuale (xm + s) fra l’intensità delle bande elettroforetiche su gel di agarosio dopo colorazione con Amido Schwarz 10B.
| Albumina | α1 |
α2 |
β |
γ |
| 67,4 + 0,8 | 2,9 + 0,3 |
9,6 + 0,6 |
6,9 + 0,7 |
12,2 + 0,7 |
8.7.3. Appendice: interpretazione dei tracciati elettroforetici
 |
|
Fig. 8.4. Quadro elettroforetico sieroproteico normale (27 anni, sesso: m.) L’esempio presentato mostra la separazione elettroforetica del siero mediante una tecnica "semimicro" che consente di evidenziare 5 frazioni: (da sinistra a destra) la banda dell’albumina, la banda α1, la banda α2, la banda β e la banda γ. La banda dell’albumina è in posizione anodica ed è quella con maggiore mobilità elettroforetica, mentre la banda γ è in posizione catodica ed è la più lenta.
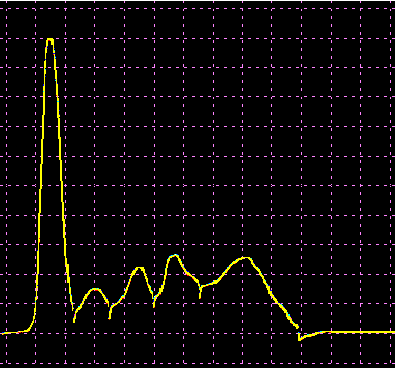 |
|
Fig. 8.5. Grave ipoalbuminemia da enteropatia essudativa (55 anni, sesso: m.) L’ipoalbuminemia grave (< 20 g/L) si riscontra in condizioni molto diverse con quadri elettroforetici in cui è preminente o il difetto di sintesi epatica o la perdita proteica attraverso i reni, l’intestino, la cute o le sierose. Quando predomina un difetto di sintesi epatica, la concentrazione totale delle sieroproteine non si discosta generalmente molto dalla norma, vi è spesso una diminuzione anche delle altre frazioni proteiche di origine epatica (α1, α2, β), mentre vi è di regola un aumento delle γ-globuline. Quando predomina la perdita proteica, la proteinemia totate è molto bassa, si osserva un forte calo delle g-globuline (se non è compensato da un aumento della loro sintesi) e, a volte, una diminuzione della sola frazione α1. Nel caso in esame non si può escludere una contemporanea compromissione della sintesi epatica dell’albumina e una stimolazione compensativa della sintesi delle immunoglobuline.
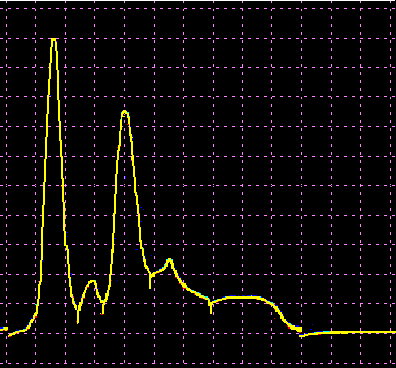 |
|
Fig. 8.6. Sindrome nefrosica (10 anni, sesso: m.) Il quadro elettroforetico è caratterizzato da una grave diminuzione dell’albumina, da una più lieve diminuzione della frazione γ e da uno spiccato incremento dell’a2-macroglobulina nella porzione anodica del picco α2. La presenza di proteine nelle urine (8 g/24 h) suffraga la diagnosi. Misure immunonefelometriche confermano che l’α2-macroglobulina è effettivamente aumentata (16 g/L; valori di riferimento: 1,5 - 3,5 g/L) e che l’aptoglobina, presente sul versante catodico del picco a2, è nei limiti della norma (2 g/L, valori di riferimento: 0,6 -2,7 g/L).
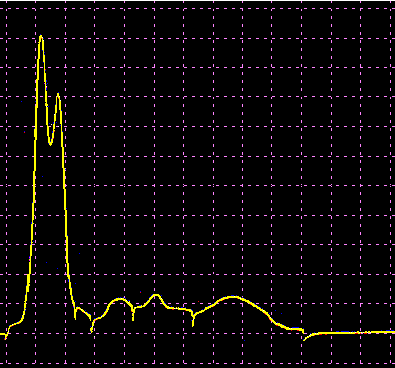 |
|
Fig. 8.7. Bisalbuminemia ereditaria (49 anni, sesso: f.) Nel soggetto eterozigote per una variante di tipo familiare è possibile evidenziare all’elettroforesi la presenza di due frazioni albuminemiche pressoché eguali: la prima con la mobilità elettroforetica dell’albumina normale, la seconda con una mobilità maggiore (variante fast) o minore (variante slow).
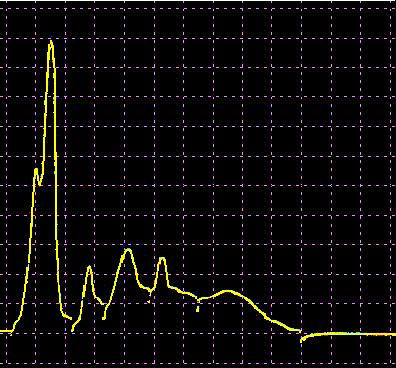 |
|
Fig. 8.8. Pseudobisalbuminemia da pseudocisti pancreatica (40 anni, sesso: m.) La transitoria comparsa di una banda albuminemica con maggiore mobilità elettroforetica può essere indotta da antibiotici (somministrazione di alte dosi di betalattamici a un soggetto con ipoalbuminemia, ad esempio in un caso di nefrosi) o dalla fistolizzazione di una pseudocisti pancreatica in cavità pleurica o peritoneale (per riassorbimento in circolo dell’albumina dell’essudato, parzialmente digerita dalle proteasi pancreatiche riversate in cavità).
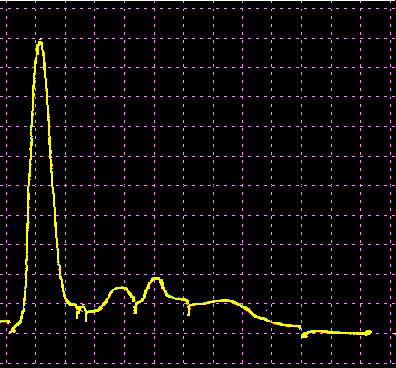 |
|
Fig. 8.9. Deficit congenito di a1-antitripsina (18 anni, sesso: m.) Il riscontro di un deficit di α1-antitripsina impone un controllo delle condizioni cliniche associate (disturbi polmonari ascrivibili ad enfisema giovanile, alterazioni epatiche) ed un allargamento delle indagini ai consanguinei.
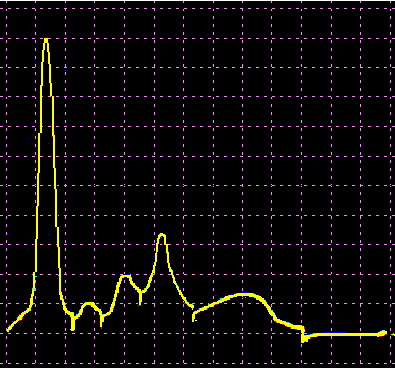 |
|
Fig. 8.10. Aumento della transferrina in corso di gravidanza (32 anni, sesso: f.) L’aumento della transferrina si evidenzia in un picco piuttosto ristretto in zona β. Le proteine che incidono sulla formazione della frazione β sono la transferrina (in posizione anodica) e la componente C3 del complermento (in posizione catodica). Su alcuni supporti, il picco β può essere influenzato anche dall’aumento delle β-lipoproteine. In zona β sono presenti, come sottofondo, anche quote (generalmente modeste) di immunoglobuline. L’aumento della transferrina è generalmente associato a carenza di ferro, cosa non rara in corso di gravidanza, quando peraltro si osserva anche un aumento delle β-lipoproteine.
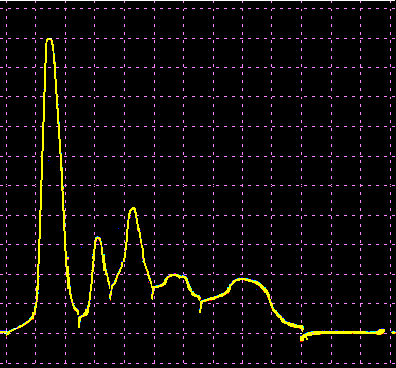 |
|
Fig. 8.11. Risposta di fase acuta in corso di pleurite (55 anni, sesso: f.) Nella risposta di fase acuta sono documentabili, mediante l’elettroforesi, l’aumento della frazione α1 e/o della frazione α2, da attribuirsi rispettivamente ad un incremento dell’α1-antitripsina e dell’aptoglobina. Questi aumenti sono di regola accompagnati da una significativa diminuzione dell’albumina, tranne che nei casi di neoplasia. Misure più accurate si ottengono mediante dosaggi immunochimici delle singole proteine.
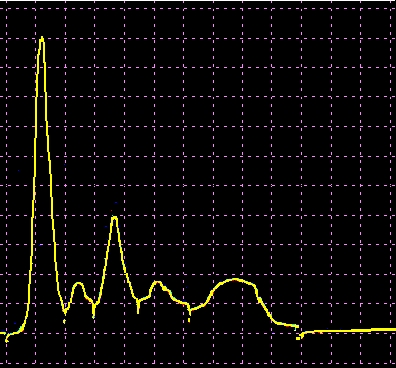 |
|
Fig. 8.12. Broncopolmonite (67 anni, sesso: f.). L’elettroforesi mostra un quadro reattivo misto caratterizzato da un aumento sia delle frazioni α1 e α2 sia della frazione γ, che rimane ben separata dal picco β. L’aumento policlonale delle immunoglobuline è indice di una reazione infiammatoria che dura da lungo tempo con eventuali episodi di riacutizzazione ed è osservabile in numerose malattie infettive croniche.
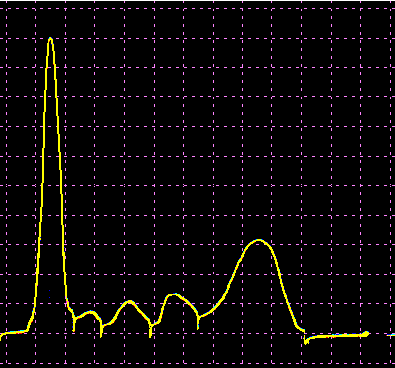 |
|
Fig. 8.13. Leishmaniosi (22 anni, sesso: m.) Il quadro elettroforetico è caratterizzato da una ipergammaglobulinemia policlonale, non associata a contemporaneo aumento delle frazioni α1 e α2, ed è osservabile in diverse malattie infettive croniche (tubercolosi, sifilide, micosi profonde, osteomielite, bronchite cronica con bronchiectasie, pielonefrite cronica, lebbra, malaria, leishmaniosi, tripanosomiasi, etc.). L’albumina è diminuita.
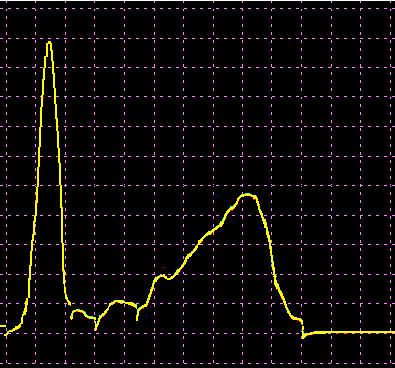 |
|
Fig. 8.14. Cirrosi epatica (41 anni, sesso:m.) Il quadro elettroforetico mostra una spiccata ipergammaglobulinemia con fusione delle frazioni β e γ. La fusione dei due picchi è dovuta essenzialmente ad un aumento delle IgA e delle IgG. Nelle cirrosi è inoltre associato un aumento delle IgM che generalmente manca nelle epatiti croniche persistenti. Il dosaggio frazionato delle immunoglobuline ha infatti dato i seguenti valori: IgA = 6,5 g/L (valori di riferimento: 1,4-3,5 g/L); IgG = 30,6 g/L (valori di riferimento: 8,0-16 g/L); IgM = 5,1 g/L (valori di riferimento: 0,5-2,0 g/L). L’ipergammaglobulinemia della cirrosi epatica è spesso accompagnata ad una diminuzione delle proteine di sintesi epatica (soprattutto l’aptoglobina e la transferrina, mentre possono risultare conservate l’α1-antitripsina e l’α2-macroglobulina).
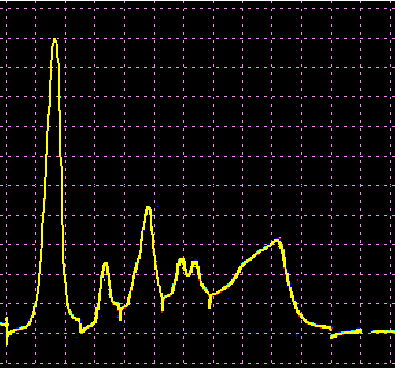 |
|
Fig. 8.15. Epatite lupoide (56 anni, sesso: f.) Il quadro elettroforetico mostra una frazione γ asimmetrica compatibile con la presenza di immunocomplessi circolanti, di componenti monoclonali o di componenti oligoclonali appartenenti ad una stessa classe o sottoclasse di immunoglobuline. La banda β mostra una parziale separazione della transferrina (in posizione anodica) dalla componente C3 del complememto (in posizione catodica).
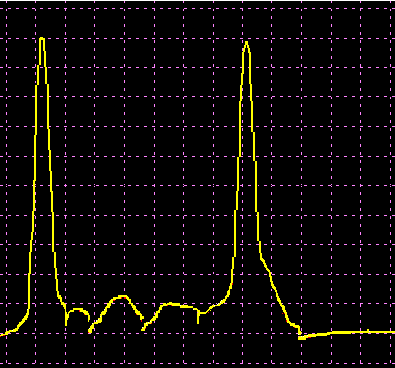 |
|
Fig. 8.16. Mieloma multiplo (68 anni, sesso: m.) Il grosso picco anomalo a base stretta nella zona γ orienta verso la diagnosi di un mieloma multiplo o di una macroglobulinemia di Waldenström, che è caratterizzata dalla presenza di immunoglobuline monoclonali di tipo IgM. E’ opportuno perciò eseguire degli esami supplementari per identificare la classe molecolare della componente anomala e documentarne l’eventuale derivazione monoclonale, dimostrando la presenza di un solo tipo di catene leggere (k o λ) sulla immunoglobulina. E’ inoltre utile eseguire una elettroforesi delle urine per accertare la presenza di immunoglobuline monoclonali o dei loro frammenti (ad esempio, le catene leggere). Nel caso in esame il dosaggio delle immunoglobuline ha dato i seguenti valori: IgA = < 0,4 g/L (valori di riferimento: 1,4-3,5 g/L); IgG = 39,0 g/L (valori di riferimento: 8,0-16 g/L); IgM = 0,3 g/L (valori di riferimento: 0,5-2,0 g/L). Mediante tecniche di immunofissazione è stata dimostrata la positività della banda in γ agli antisieri IgG e k. L’elettroforesi delle urine ha evidenziato una piccola banda monoclonale in zona γ.
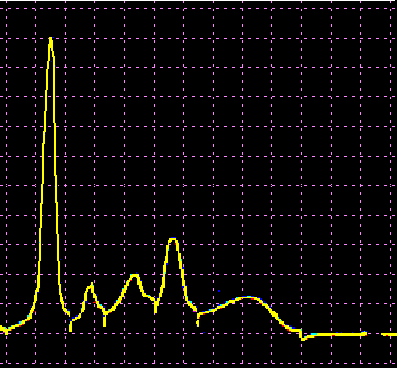 |
|
Fig. 8.18. Linfoma non-Hodgkin (63 anni, sesso: m.) L’aumento non cospicuo della frazione β ha ingenerato il sospetto della presenza di una componente monoclonale per l’intensità e la compatezza della banda all’ispezione visiva della lastrina. Il modico aumento delle α1 e delle α2 depone per una flogosi acuta. Il dosaggio delle immunoglobuline ha dato i seguenti valori: IgA < 0,4 g/L (valori di riferimento: 1,4-3,5 g/L); IgG = 11,9 g/L (valori di riferimento: 8,0-16 g/L); IgM = 15,0 g/L (valori di riferimento: 0,5-2,0 g/L). La presenza di una componente monoclonale è stata confermata mediante immunofissazione, dimostrando la positività della banda elettroforetica in β agli antisieri IgM e k. L’elettroforesi delle urine non ha evidenziato bande monoclonali..
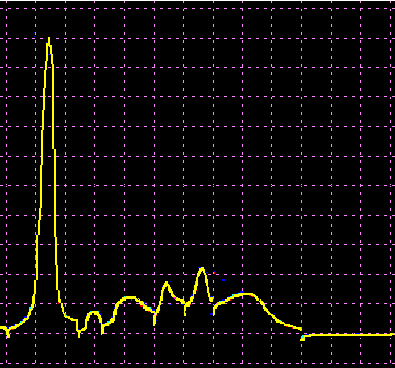 |
|
Fig. 8.18. Gammapatia monoclonale (65 anni, sesso: f.) Un picco monoclonale può essere evidenziato in affezioni benigne o in processi morbosi di varia natura (adenocarcinoma del retto, tubercolosi, lue, endocardite batterica, colecistite ricorrente, epatopatie, etc.). Il picco è generalmente meno sviluppato che nei mielomi e nella malattia di Waldenström e può essere presente in zona γ o in zone più anodiche, sovrapponendosi alle bande α2 o β. Nel caso in esame il picco anomalo è in zona φ, intermedia tra le bande β e γ, dove migra il fibrinogeno del plasma. Il dosaggio delle immunoglobuline ha dato i seguenti valori: IgA = 9,5 g/L (valori di riferimento: 1,4-3,5 g/L); IgG = 12,5 g/L (valori di riferimento: 8,0-16 g/L); IgM = 0,95 g/L (valori di riferimento: 0,5-2,0 g/L). L’immunofissazione ha dimostrato una banda in φ positiva agli antisieri IgA e λ. L’elettroforesi delle urine ha evidenziato una piccola banda monoclonale in zona φ.
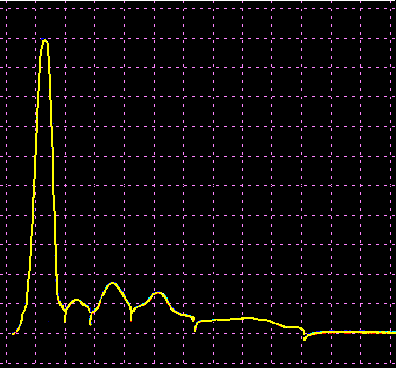 |
|
Fig. 8.19. Ipogammaglobulinemia in corso di leucemia linfatica cronica (58 anni, sesso: f.) Un decremento nella sintesi di gammaglobulinemie può essere osservato nel corso di neoplasie di varia origine, di infezioni e di trattamento con immunosoppressori o in caso di deficit congeniti selettivi . Una carenza dovuta ad eccessiva perdita di immunoglobuline si riscontra nella sindrome nefrosica e nelle gastroenteropatie proteinodisperdenti, mentre un accellerato catabolismo di questa frazione proteica si ha nella ipoproteinemia familiare e nella distrofia miotonica. La frazione γ è di norma ridotta nei bambini di età inferiore ai 6 anni.
| aggiornamento: 04/10/14 |