|
|
Costantino Salerno |
|
|
Appunti di Biochimica Clinica |
|
|
10. ORMONI
Gli ormoni sono composti di natura organica che sono sintetizzati in particolari tessuti (usualmente specifiche ghiandole prive di dotti escretori) e sono distribuiti, tramite il circolo sanguigno, in altri distretti corporei dove producono effetti regolatori.
Sono ormoni in senso stretto (o ghiandolari) quelle sostanze biologicamente attive che sono prodotte nelle ghiandole endocrine e sono considerate come tali l’ipofisi, la tiroide, le ghiandole paratiroidi, le ghiandole surrenali (corticale e midollare), le gonadi e le isole pancreatiche. Sono definiti ormoni tessutali (o istiormoni) quei composti chimici con proprietà analoghe a quelle degli ormoni, ma che non vengono prodotti in ben definiti organi endocrini. Rientrano in questo gruppo, ad esempio, la secretina e la colecistochinina prodotte a livello intestinale, la serotonina prodotta dalle cellule argentaffini gastrointestinali, i peptidi natriuretici prodotti a livello cardiaco, la 1,25-diidrossi vitamina D prodotta a livello dei tubuli renali prossimali, l’eritropoietina e l’angiotensina II che derivano da precursori già presenti nel plasma.
Bisogna tuttavia ricordare che alcuni ormoni propriamente detti sono sintetizzati, oltre che a livello di ghiandole endocrine, anche in strutture diverse: questo può valere, ad esempio, per la noradrenalina che viene liberata sia dalle cellule della midollare surrenale che dalle terminazioni nervose simpatiche, per la gastrina che è prodotta sia nelle isole pancreatiche che a livello della mucosa pilorica e duodenale e per il glucagone che viene sintetizzato prevalentemente nelle cellule A delle isole pancreatiche, ma in parte anche in analoghe cellule disseminate nella mucosa del fondo dello stomaco.
Infine, sono definiti parormoni quelle sostanze che, pur non possedendo uno o più requisiti convenzionalmente richiesti per la definizione di ormone, esplicano un controllo locale modificando l’equilibrio chimico dell’ambiente interno od esterno alle cellule, come ad esempio le prostaglandine ed i fattori di crescita nervoso (Nerve Growth Factor, NGF) ed epidermico (Epidermal Growth Factor, EGF).
Per una completa ed approfondita trattazione dell’argomento, si rimanda il lettore a trattati specialistici di endocrinologia.
10.1. ADENOIPOFISI E SISTEMA IPOTALAMICO-NEUROIPOFISARIO
L’ipofisi è costituita (1) da un lobo anteriore che si continua in alto con il processo linguiforme, che a sua volta avvolge il peduncolo e ricopre anteriormente l’eminenza mediana e (2) da un lobo posteriore. Fra il lobo anteriore e quello posteriore si osserva, nel feto e nel neonato, una sottile fessura ripiena di colloide, la cui parete posteriore costituisce il lobo intermedio, struttura non più riconoscibile come parte a sé stante nel soggetto adulto.
Il lobo anteriore, il lobo intermedio e il processo linguiforme costituiscono
l’adenoipofisi, di cui entra a far
parte anche una struttura rudimentale (l’ipofisi faringea)
situata nella sottomucosa della volta faringea e completamente
separata dalle restanti strutture ipofisarie a mezzo del corpo
dello sfenoide. Il lobo posteriore, il peduncolo ipofisario e l’eminenza
mediana costituiscono il sistema ipotalamico-neuroipofisario.
L’eminenza mediana fa parte (assieme alla ghiandola pineale, all’organum
vasculosum della lamina terminale, all’organo subfornicale e all’organo subcommissurale) di una serie di strutture impari associate al terzo ventricolo
prive di barriera emato-encefalica, che possono servire da finestra nel
trasporto di sostanze dal circolo sanguigno al cervello. La ghiandola pineale
sintetizza la melatonina (5-metossi-N-acetilserotonina) a partire dalla
serotonina
![]() ; la produzione di melatonina è inibita dalla luce, mentre è massima
di notte contribuendo così al mantenimento del ritmo circandiano veglia/sonno e all’abbassamento notturno della temperatura
corporea (Fig. 10.1).
; la produzione di melatonina è inibita dalla luce, mentre è massima
di notte contribuendo così al mantenimento del ritmo circandiano veglia/sonno e all’abbassamento notturno della temperatura
corporea (Fig. 10.1).
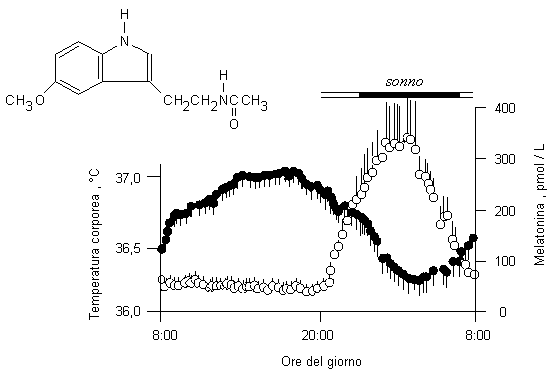
Fig. 10.1. Ritmo circandiano di increazione della melatonina. Vi è una evidente correlazione tra l’incremento della melatonina è l’abbassamento della temperatura corporea. Gli eventi precedono di circa tre ore l’inizio del sonno. La concentrazione della melatonina nel siero è indicata con i cerchi vuoti. La temperatura corporea è indicata con i cerchi pieni. L’errore standard delle misure è indicato dalle barre verticali. L’inserto mostra la formula di struttura della melatonina.
L’adenoipofisi è costituita da isole cellulari solide, fittamente stipate fra loro, circondate da sottili tralci di tessuto connettivo fibrillare in cui decorrono i vasi sanguigni e con al centro, a volte, una piccola quantità di sostanza colloide in posizione extracellulare. Nella maggior parte delle cellule ghiandolari dell’adenoipofisi sono presenti granuli citoplasmatici con caratteristiche istochimiche diverse a seconda del tipo di cellula. La sintesi, l’accumulo e la liberazione dei differenti ormoni sono strettamente correlati con la formazione e lo scarico dei granuli citoplasmatici e, a sua volta, la natura dell’ormone secreto da ogni tipo cellulare è intimamente correlato con la natura chimica dei granuli secretori contenuti nel citoplasma. Il lobo posteriore dell’ipofisi è formato da un fitto intreccio di fibre nervose amieliniche e dalle loro terminazioni a fondo cieco, presentanti spesso un rigonfiamento a clava (corpi di Herring), che costituiscono la parte più distale degli assoni delle cellule dei nuclei sopraottico e paraventriclorari, giunti all’ipofisi attraverso il peduncolo. Tra le fibre nervose sono presenti cellule gliari (pituiciti) di forma e dimensioni varie, vasi sanguigni piuttosto scarsi, una certa quota di tessuto connettivo fibrillare e un pigmento che, assente in età giovanile, si accumula, in parte in sede extracellulare, col procedere degli anni. Nei punti di contatto tra lobo anteriore e posteriore si riscontrano inoltre alcune isole di cellule con caratteri sovrapponibili a quelli delle cellule cianofile del lobo anteriore (cellule zeta). Nel secondo decennio di vita queste cellule aumentano di numero e tendono a guadagnare gradualmente le porzioni più posteriori della ghiandola (invasione basofila del lobo posteriore).
L’ipofisi propriamente detta è irrorata dalle arterie ipofisarie, rami della carotide interna, mentre il peduncolo e l’eminenza mediana sono riforniti di sangue arterioso che deriva da sottili rami provenienti dal circolo di Willis. Dalle reti capillari si formano rami venosi che presentano un comportamento diverso a seconda del settore ipofisario in esame: mentre a livello dell’ipofisi propriamente detta gli effluenti venosi dei due lobi, anteriore e posteriore, sono in connessione direttamente con le vene sistemiche, i rami venosi provenienti dalle strutture nervose dell’eminenza mediana penetrano nelle strutture ghiandolari del processo linguiforme e del lobo anteriore, costituendo il sistema portale ipofisario e sfioccandosi in una nuova rete capillare. L’importanza funzionale di questa peculiare situazione vascolare risiede nel fatto che sostanze defluenti con il sangue venoso dalle strutture nervose dell’eminenza mediana possono raggiungere le strutture ghiandolari del lobo anteriore ed esercitare per via umorale la loro azione. Questo meccanismo non risulta invece possibile tra il lobo posteriore e il lobo anteriore che sono separati, come si è detto, dai residui della fessura ipofisaria e sono privi di un interscambio di sangue.
All’adenoipofisi viene attribuita la funzione di
produrre almeno sei ormoni, di cui tre di natura proteica, la
somatotropina (hGH), la
corticotropina (ACTH) e la
prolattina (hPrL),
e tre di natura glicoproteica, l’ormone tireotropo (TSH), l’ormone
follicolo-stimolante (FSH) e l’ormone luteinizzante (LH)
![]() . L’increzione
degli ormoni adenoipofisari è controllata da
fattori di rilascio o di inibizione liberati nel
circolo portale ipofisario dalle terminazioni nervose di neuroni ipotalamici,
che risentono, a loro volta, degli effetti
di neurotrasmettitori prodotti da altri neuroni situati a livello
ipotalamico e nelle regioni sovrastanti.
. L’increzione
degli ormoni adenoipofisari è controllata da
fattori di rilascio o di inibizione liberati nel
circolo portale ipofisario dalle terminazioni nervose di neuroni ipotalamici,
che risentono, a loro volta, degli effetti
di neurotrasmettitori prodotti da altri neuroni situati a livello
ipotalamico e nelle regioni sovrastanti.
Il sistema ipotalamico-neuroipofisario produce due ormoni peptidici sicuramente distinti, l’ossitocina e la vasopressina (arginine vasopressin, AVP), che differiscono fra loro soltanto per due aminoacidi (Fig. 10.2). Questi ormoni e le neurofisine, che servono da proteine di trasporto dell’ossitocina e della vasopressina, derivano dalla parziale idrolisi di due glicoproteine (la pro-pressofisina e la pro-oxifisina) presenti nel tessuto neuroipofisario.
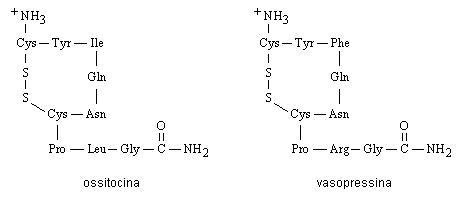
Fig. 10.2. Ormoni prodotti dal sistema ipotalamico-neuroipofisario
L’ossitocina stimola il miometrio a contrarsi durante la fase espulsiva del parto sia direttamente sia provocando il rilascio di prostaglandine ed assicurando altresì l’emostasi durante il secondamento, ma non induce di per sé il parto nella donna. L’aumento della concentrazione plasmatica di ossitocina è provocato per via riflessa dalla distensione delle pareti vaginali durante la fase terminale del parto. Tuttavia, anche in assenza di un aumento del livello di ossitocina nel plasma, il miometrio viene stimolato in ogni caso a contrarsi in quanto gli estrogeni inducono un aumento del numero di recettori per l’ossitocina nel miometrio durante la gravidanza e, in particolare, in prossimità del parto.
Durante il periodo dell’allattamento, l’increzione di ossitocina è indotta nella donna da stimoli psicogeni o mediante una stimolazione delle terminazioni nervose dei capezzoli attraverso la suzione, mentre episodi di paura, collera o stress possono inibire il rilascio dell’ormone. L’ossitocina induce, a sua volta, la contrazione delle cellule mioepiteliali situate lungo i dotti lattiferi e attorno agli alveoli della ghiandola mammaria agevolando la fuoriuscita del latte (Fig. 10.3).
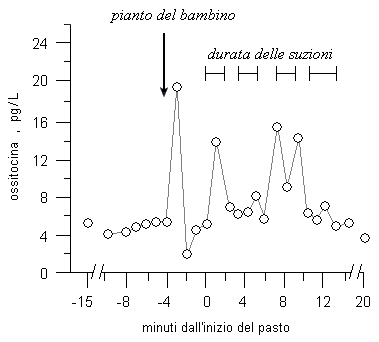
Fig. 10.3. Variazioni della concentrazione plasmatica di ossitocina in una donna che allatta un bambino. L’increzione dell’ossitocina è indotta sia da stimoli psicogeni sia per via riflessa dalla suzione del lattante.
La vasopressina è coinvolta nei meccanismi omeostatici che
regolano (1) l’osmolarità del sangue, (2) la volemia e (3)
l’increzione della corticotropina (vedi Par. 10.5.1a)
![]() . L’ormone è un
potente vasocostrittore e svolge una azione antidiuretica favorendo il
riassorbimento dell’acqua a livello renale (vedi
Par. 11). L’osmolarità del sangue è monitorata da osmorecettori
localizzati nell’ipotalamo anteriore ed è mantenuta a un valore prossimo a 285 mOsm/kg con uno scarto inferiore a +1,8%. La diminuzione della
pressione sanguigna conseguente ad una riduzione della volemia attiva i
barocettori localizzati nell’atrio sinistro, nella vena cava e nelle vene
polmonari che trasmettono il segnale per via riflessa all’ipotalamo nonché i barocettori renali che inducono un aumento di angiotensina II nel sangue
attraverso l’attivazione del sistema renina-angiotensina (vedi
Par. 10.5.1b). A sua volta, l’angiotensina II stimola la sintesi dell’aldosterone,
provoca una vasocostrizione periferica e, superata la barriera emato-encefalica,
stimola direttamente l’ipotalamo.
. L’ormone è un
potente vasocostrittore e svolge una azione antidiuretica favorendo il
riassorbimento dell’acqua a livello renale (vedi
Par. 11). L’osmolarità del sangue è monitorata da osmorecettori
localizzati nell’ipotalamo anteriore ed è mantenuta a un valore prossimo a 285 mOsm/kg con uno scarto inferiore a +1,8%. La diminuzione della
pressione sanguigna conseguente ad una riduzione della volemia attiva i
barocettori localizzati nell’atrio sinistro, nella vena cava e nelle vene
polmonari che trasmettono il segnale per via riflessa all’ipotalamo nonché i barocettori renali che inducono un aumento di angiotensina II nel sangue
attraverso l’attivazione del sistema renina-angiotensina (vedi
Par. 10.5.1b). A sua volta, l’angiotensina II stimola la sintesi dell’aldosterone,
provoca una vasocostrizione periferica e, superata la barriera emato-encefalica,
stimola direttamente l’ipotalamo.
Alterazioni del sistema omeostatico in cui è coinvolta la
vasopressina portano ad una eccessiva perdita o ritenzione di acqua
![]() .
Il diabete insipido è caratterizzato da poliuria e polidipsia, è
conseguente ad un deficit di increzione di vasopressina e può essere dovuto a
lesioni o compressioni della regione ipotalamica o (più raramente) ad
alterazioni congenite. Poiché il senso della sete è di norma conservato nei
pazienti con diabete insipido, generalmente non si riscontra una
alterazione dell’osmolarità del plasma. La
malattia può essere associata a disturbi della vista in caso di lesioni del chiasma
ottico o ad ipopituitarismo ed elevati livelli ematici di prolattina (vedi Par.
10.3) in caso di interruzione del tratto neuroipofisario.
.
Il diabete insipido è caratterizzato da poliuria e polidipsia, è
conseguente ad un deficit di increzione di vasopressina e può essere dovuto a
lesioni o compressioni della regione ipotalamica o (più raramente) ad
alterazioni congenite. Poiché il senso della sete è di norma conservato nei
pazienti con diabete insipido, generalmente non si riscontra una
alterazione dell’osmolarità del plasma. La
malattia può essere associata a disturbi della vista in caso di lesioni del chiasma
ottico o ad ipopituitarismo ed elevati livelli ematici di prolattina (vedi Par.
10.3) in caso di interruzione del tratto neuroipofisario.
10.2. ORMONE DELLA CRESCITA
La somatotropina (ormone somatotropo o della crescita; hGH) è prodotta dalle cellule somatotrope (cellule alfa) che rappresentano circa il 50% delle cellule ipofisarie e sono localizzate prevalentemente nelle parti laterali del lobo anteriore. All’interno di queste cellule, l’ormone si trova accumulato in granuli rotondeggianti di 250-500 nm.
L’ormone presente nell’ipofisi e nel plasma è costituito da un gruppo di proteine simili tra loro, ma di diversa struttura e peso molecolare. La più comune forma molecolare, che rappresenta circa il 70% dell’ormone circolante, è una proteina di 22 kDa composta da una singola catena di 191 aminoacidi stabilizzata da due ponti disolfuro. Questa proteina deriva da un preormone (pre-hGH) di 24 kDa con una catena aggiuntiva di 26 aminoacidi all’estremità amino-terminale, che rappresenta una sequenza segnale per il trasporto della proteina all’interno del reticolo endoplasmatico. Circa il 20% dell’ormone circolante è costituito da una molecola più piccola con una massa molecolare di 20 kDa, che è priva del frammento polipeptidico dall’aminoacido 32 al 46. Un riarrangiamento del prodotto di trascrizione con eliminazione del frammento polipeptidico dall’aminoacido 32 al 71 porta invece alla formazione di una proteina di 17,5 kDa. Altre frazioni sono acetilate o deaminate o formano dimeri privi di attività biologica, ma con le proprietà antigeniche dell’ormone nativo. Sul piano strutturale, la somatotropina è molto simile alla somatomammotropina corionica (Par. 10.7.1) ed alla prolattina (Par. 10.3), possedendo anche alcune azioni in comune con queste due molecole.
L’increzione di somatotropina avviene in modo discontinuo e soprattutto durante le prime ore del sonno (vedi Fig. 10.4) ed è controllata da due polipeptidi interdipendenti, l’ormone rilasciante la somatotropina (GHRH, 44 aminoacidi) e la somatostatina (SMS, 14 aminoacidi). Quando giunge in periferia, la somatotropina viene metabolizzata in gran parte dal fegato e dal rene. Gli effetti della somatotropina sull’accrescimento somatico sono mediati da fattori di crescita insulino-simili, la somatomedina C (IGF-I) e la somatomedina A (IGF-II). La somatomedina C è un polipeptide di 8 kDa simile alla proinsulina e strettamente dipendente dalla somatotropina per quanto riguarda la sua concentrazione plasmatica. Somatomedina A è molto simile alla somatomedina C, ma ha una attività più spiccatamente insulinosimile ed è meno dipendente dalla somatotropina. Le somatomedine sono, a loro volta, veicolate nel sangue da proteine di trasporto specifiche (IGF-binding protein 1 e 3) sintetizzate a livello del fegato e dell’endometrio e capaci di modulare l’attività biologica delle somatomedine stesse.
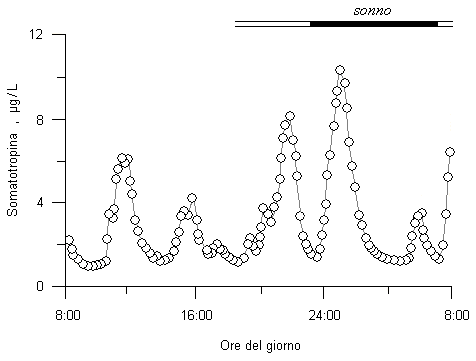
Fig. 10.4. Variazione della concentrazione plasmatica di somatotropina nel corso di una giornata. L’ormone viene increto in modo discontinuo e soprattutto durante le prime ore del sonno.
La somatotropina non è essenziale per lo sviluppo del feto.
Il suo deficit durante l’età pediatrica è causa di
bassa statura e può associarsi a ipoglicemia
![]() .
Un eccesso di somatotropina nel bambino è estremamente più raro
ed è causa di
gigantismo. In età adulta, una sovraproduzione
dell’ormone determina l’acromegalia. Nella maggior
parte dei casi questa malattia è dovuta ad un adenoma ipofisario
secernente l’ormone, più raramente è legata ad una
iperincrezione di GHRH che può avere origine da neoplasie
ipotalamiche o, più frequentemenete, da tumori extraipofisari (carcinoidi
bronchiali, tumori gastrointestinali e pancreatici). Estremamente
rara è l’acromegalia da produzione ectopica di
somatotropina in caso di tumore pancreatico.
.
Un eccesso di somatotropina nel bambino è estremamente più raro
ed è causa di
gigantismo. In età adulta, una sovraproduzione
dell’ormone determina l’acromegalia. Nella maggior
parte dei casi questa malattia è dovuta ad un adenoma ipofisario
secernente l’ormone, più raramente è legata ad una
iperincrezione di GHRH che può avere origine da neoplasie
ipotalamiche o, più frequentemenete, da tumori extraipofisari (carcinoidi
bronchiali, tumori gastrointestinali e pancreatici). Estremamente
rara è l’acromegalia da produzione ectopica di
somatotropina in caso di tumore pancreatico.
10.2.1. Metodi di determinazione
I saggi biologici per la determinazione della somatotropina sono usati per dosare l’attività di peptidi naturali o sintetici e per la calibrazione di soluzioni standard. Essi si basano sulla valutazione di un alcuni cambiamenti metabolici indotti dall’ormone in ratti ipofisectomizzati. Micrometodi adatti a determinazioni su piccoli volumi consistono nel misurare l’incremento di crescita di linee cellulari in coltura in seguito all’aggiunta dell’ormone. Altri metodi consistono nel determinare il legame dell’ormone a preparazioni di recettori ottenute da fegato di ratto, sfruttando la competizione con una preparazione standard di somatotropina marcata con 125-I.
Nella routine clinica l’ormone è generalmente
dosato mediante metodi radioimmunologici (RIA) basati sulla
competizione, rispetto ad un numero limitato di siti anticorpali,
del campione in esame con una preparazione standard
radioattiva. Il complesso antigene-anticorpo viene isolato
mediante precipitazione in polietilen glicol o in miscele di
etanolo e solfato d’ammonio o mediante l’utilizzo di un
secondo anticorpo insolubilizzato
![]() . L’eccesso di materiale
radioattivo viene quindi allontanato dall’aggregato
macromolecolare mediante lavaggio.
. L’eccesso di materiale
radioattivo viene quindi allontanato dall’aggregato
macromolecolare mediante lavaggio.
10.2.2. Preparazione del campione ed intervalli di riferimento
Le indagini ormonali comprendono la valutazione della increzione spontanea e dopo stimolazione della somatotropina ed il dosaggio della somatomedina C e di una sua proteina di legame (IGF-binding protein 3).
Lo studio della increzione ormonale spontanea non ha valore diagnostico nell’adulto, in quanto è stata documentata una ampia sovrapposizione dei valori normali con quelli patologici. Nel bambino, lo studio della increzione spontanea di somatotropina è indispensabile per identificare l’esistenza di una disfunzione neurosecretoria caratterizzata da ridotta concentrazione di hGH e somatomedina C e da una normale risposta ai test di stimolazione.
Al fine di valutare la capacità incretoria dell’ipofisi,
sono stati standardizzati alcuni test di stimolazione
con insulina, arginina, clonidina, L-dopa o glucagone che
prevedono, come minima risposta normale, un valore di
somatotropina sierica di 7-10 μg/L. L’alta
frequenza di risposte falsamente negative limita tuttavia l’affidabilità
diagnostica di queste prove. Maggiore valore è attribuito alla
stimolazione con GHRH esogeno quando questa è preceduta dalla
somministrazione di piridostigmina (un agonista colinergico
indiretto) o di arginina ![]() . In questi casi la
risposta normale è di 20 mg/L nel
bambino e di 16,5 μg/L nell’adulto.
Il test con arginina e GHRH conserva una elevata
affidabilità diagnostica per tutto il corso della vita, mentre l’effetto
potenziante della piridostigmina si riduce nell’età senile
in conseguenza della riduzione dell’attività colinergica a
livello del sistema nervoso centrale, propria di questa età.
. In questi casi la
risposta normale è di 20 mg/L nel
bambino e di 16,5 μg/L nell’adulto.
Il test con arginina e GHRH conserva una elevata
affidabilità diagnostica per tutto il corso della vita, mentre l’effetto
potenziante della piridostigmina si riduce nell’età senile
in conseguenza della riduzione dell’attività colinergica a
livello del sistema nervoso centrale, propria di questa età.
La determinazione della concentrazione sierica della
somatomedina C è una delle prime indagini diagnostiche richieste
in caso di sospetto deficit di somatotropina. La concentrazione
sierica della somatomedina C, che si aggira nel soggetto normale
attorno a 200 μg/L
![]() ,
dipende tuttavia dallo stato nutrizionale e dalle condizioni
metaboliche del paziente. L’affidabilità diagnostica della
determinazione di questo fattore è assai variabile in rapporto
all’età, in considerazione della riduzione fisiologica della somatomedina C nel corso della vita. Nei rari casi di
increzione
di somatotropina inattiva, la somministrazione di somatotropina
esogena determina un innalzamento dei bassi livelli di
somatomedina C.
,
dipende tuttavia dallo stato nutrizionale e dalle condizioni
metaboliche del paziente. L’affidabilità diagnostica della
determinazione di questo fattore è assai variabile in rapporto
all’età, in considerazione della riduzione fisiologica della somatomedina C nel corso della vita. Nei rari casi di
increzione
di somatotropina inattiva, la somministrazione di somatotropina
esogena determina un innalzamento dei bassi livelli di
somatomedina C.
La determinazione della IGF-binding protein 3 non sembra portare ulteriori elementi utili per la diagnosi, anche in considerazione del fatto che questa proteina risulta essere un marker dello stato incretorio della somatotropina meno sensibile della somatomedina C.
10.3. PROLATTINA
Le cellule lattotrope dell’adenoipofisi, che producono la prolattina (ormone luteotropo; hPrL), costituiscono circa il 10-25% della ghiandola in condizioni normali e il 70% in corso di gravidanza. La prolattina è una proteina di 198 aminoacidi (23 kDa) stabilizzata da tre ponti disolfuro e strutturalmente simile all’ormone somatotropo e alla somatomammotropina corionica (vedi Par. 10.2). La prolattina è presente nel plasma sia in forma monomerica, sia come dimero o trimero (big prolactin), sia come un aggregato ad alto peso molecolare (big big prolactin). Queste forme molecolari hanno la medesima immunoreattività, ma diversa attività biologica.
La increzione ipofisaria di prolattina, che avviene in maniera episodica, aumenta in seguito a stress fisici o psichici, durante il sonno e, in particolare modo, nel corso della gravidanza e durante l’allattamento. La increzione di prolattina è controllata principalmente dalla dopamina ipotalamica, che raggiunge le cellule lattotrope attraverso il sistema portale ipofisario ed agisce come un fattore inibente la increzione attraverso un meccanismo di retroreazione (feed-back) controllato dalla prolattinemia. L’aumento dei livelli di prolattina che si osserva durante la suzione del latte è probabilmente legato all’azione del Vasoactive Intestinal Peptide (VIP), del Thyrotropin-Releasing Hormone (TRH; vedi Par. 10.4) e di meccanismi serotoninergici non ancora ben definiti. La increzione di prolattina è inoltre favorita dalla serotonina e dagli estrogeni, mentre l’acido g-aminobutirrico (GABA) ha un effetto di stimolazione a livello centrale e di inibizione a livello ipofisario.
La prolattina agisce direttamente sulla ghiandola mammaria, adeguatamente preparata dagli estrogeni e dal progesterone (come accade in gravidanza), favorendo l’inizio e il mantenimento della lattazione. Recettori per la prolattina sono presenti nelle cellule della granulosa e nel testicolo. Tuttavia, mentre si è visto che la prolattina inibisce in vitro la steroidogenesi follicolare, non è stata dimostrata un’azione diretta della prolattina sul testicolo.
Una eccessiva increzione di prolattina (iperprolattinemia) è
un fenomeno patologico piuttosto comune nella donna, dove è causa
frequente di amenorrea (è alla base del 10-40% di tutte le
amenorree
![]() ),
e si associa a galattorrea nel 30-80% dei casi. Nel maschio l’iperprolattinemia
è un evento più raro e si associa spesso ad impotenza. La causa
più frequente è un adenoma ipofisario secernente la prolattina
(i prolattinomi costituiscono circa il 50% di tutti i tumori
ipofisari). In rari casi si può osservare una produzione
ectopica di prolattina nel cancro del polmone o del rene. Altre
cause sono una lesione del peduncolo ipofisario con
compromissione del circolo portale, un aumento dei fattori che
favoriscono la increzione di prolattina (estrogeni, TRH nell’ipotiroidismo
primitivo), l’assunzione di farmaci antidopaminergici (fenotiazine,
butirrofenoni, metoclopramide) o depletori di dopamine (metildopa,
reserpina), e una ridotta clearance della prolattina
come nella insufficienza renale.
),
e si associa a galattorrea nel 30-80% dei casi. Nel maschio l’iperprolattinemia
è un evento più raro e si associa spesso ad impotenza. La causa
più frequente è un adenoma ipofisario secernente la prolattina
(i prolattinomi costituiscono circa il 50% di tutti i tumori
ipofisari). In rari casi si può osservare una produzione
ectopica di prolattina nel cancro del polmone o del rene. Altre
cause sono una lesione del peduncolo ipofisario con
compromissione del circolo portale, un aumento dei fattori che
favoriscono la increzione di prolattina (estrogeni, TRH nell’ipotiroidismo
primitivo), l’assunzione di farmaci antidopaminergici (fenotiazine,
butirrofenoni, metoclopramide) o depletori di dopamine (metildopa,
reserpina), e una ridotta clearance della prolattina
come nella insufficienza renale.
10.3.1. Metodi di determinazione
I saggi biologici per la determinazione della prolattina sono generalmente poco sensibili per essere utili dal punto di vista clinico. Un metodo molto sensibile sfrutta la proprietà della prolattina di stimolare la moltiplicazione delle cellule in coltura, ma è necessario in tal caso aggiungere un siero anti-somatotropina per neutralizzare quest’ultimo ormone (vedi Par.10.2.1). I metodi di dosaggio comunemente adoperati nella pratica clinica sono radioimmunologici (RIA) e sono basati sulla competizione, rispetto ad un numero limitato di siti anticorpali, del campione in esame con una preparazione ormonale standard radioattiva.
10.3.2. Preparazione del campione ed intervalli di riferimento
Poiché la prolattina aumenta in seguito a stress e durante il sonno, il paziente deve essere in condizioni di riposo e sveglio da almeno 2 ore. E’ inoltre consigliabile prelevare diversi campioni ad intervalli di 6-18 minuti in quanto la increzione dell’ormone è discontinua. Il campione di siero può essere conservato ghiacciato, ma bisogna evitare frequenti congelamenti e scongelamenti che potrebbero denaturare la proteina.
La concentrazione di prolattina nel plasma è più alta nella donna (5-25 μg/L) che nell’uomo (5-15 μg/L), aumenta durante la gravidanza fino a raggiungere, al termine, un valore di 100-300 μg/L, cala dopo il parto, ma raggiunge valori altissimi durante la suzione.
I vari test funzionali di stimolo (con TRH, metoclopramide, clorpromazina) o di inibizione (con L-DOPA, bromocriptina) proposti per distinguere le iperprolattinemie funzionali da quelle legate ad adenomi ipofisari non danno risultati tali da consentire in tutti i pazienti una sicura precisazione diagnostica e pertanto hanno una importanza clinica modesta.
10.4. ORMONI TIROIDEI
Il parenchima tiroideo è costituito da follicoli sferoidali
di grandezza variabile, delimitati da un epitelio
monostratificato in stretto rapporto con la parete dei capillari.
I follicoli contengono al loro interno una sostanza omogenea
detta colloide costituita essenzialmente da
tireoglobulina, una glicoproteina di 660 kDa (contenente
numerosi residui di tirosina) che è sintetizzata e secreta per
esocitosi dalle cellule dell’epitelio follicolare e che può
essere considerata il precursore degli ormoni tiroidei iodinati.
Lo ioduro è captato dal sangue contro un gradiente di
concentrazione per mezzo di una pompa specifica. L’ossidazione
dello ioduro, la iodinazione dei residui di tirosina della
tireoglobulina e l’accoppiamento di due gruppi
iodotirosinici all’interno della molecola della
tireoglobulina con produzione di iodotironine sono tutti processi
catalizzati dalla perossidasi tiroidea. La tireoglobulina,
riassorbita dalle cellule follicolari, va incontro ad un processo
di proteolisi che porta alla liberazione di tetraiodotironina (tiroxina
o T4), triiodotironina (T3) e iodotirosine. Queste ultime vengono
deiodinate e lo iodio viene riutilizzato per una nuova sintesi,
mentre la T3 e la T4 attraversano la membrana basale del
follicolo e si riversano in circolo
![]() .
.
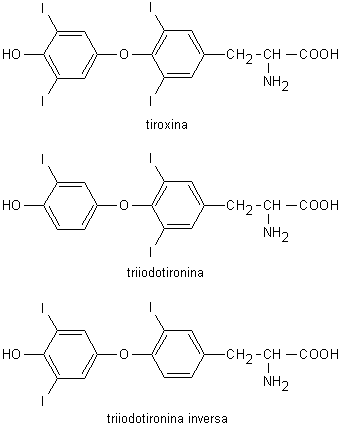
Fig. 10.5. Ormoni tiroidei
Nel plasma, le iodotironine si legano in ordine decrescente di affinità alla globulina legante la tiroxina (Thyroxine-Binding Globulin, TBG), alla prealbumina (Par. 8.1.2) e all’albumina (Par. 8.1.1). La globulina legante la tiroxina, una glicoproteina di 55 kDa di origine epatica e mobilità elettroforetica compresa tra le a1 e le a2 globuline, è il principale e più specifico trasportatore degli ormoni tiroidei. Solo lo 0,04% della T4 e lo 0,4% della T3 sono presenti nel plasma in forma libera (la T4 è circa 50 volte più concentrata della T3, ma ha anche una maggiore affinità per le proteine di trasporto).
La T4 può essere considerata un precursore della T3 in quanto
è quest’ultima molecola che media la maggior parte delle
azioni biologiche degli ormoni tiroidei legandosi a recettori
specifici ed agendo principalmente a livello dell’espressione
dei geni. Una gran parte della T3 circolante deriva dalla
deiodinazione della T4 catalizzata nel fegato e nel rene dalla
deiodinasi tipo I, un enzima selenio-dipendente capace di
staccare lo iodio sia in posizione 5’ (dando origine alla T3),
sia in posizione 5 (dando origine alla triiodotironina inversa
inattiva, rT3). La deiodinasi tipo II, presente nelle cellule
tireotrope dell’ipofisi, nella corteccia cerebrale e nel
tessuto adiposo bruno ![]() è invece in grado di staccare
esclusivamente lo iodio in posizione 5’ formando così il
solo derivato attivo. La T3 e la T4 sono degradate mediante
deaminazione, decarbossilazione ossidativa e deiodinazione. La
formazione di glicuronati e solfati permette l’eliminazione
di questi composti per via renale e biliare.
è invece in grado di staccare
esclusivamente lo iodio in posizione 5’ formando così il
solo derivato attivo. La T3 e la T4 sono degradate mediante
deaminazione, decarbossilazione ossidativa e deiodinazione. La
formazione di glicuronati e solfati permette l’eliminazione
di questi composti per via renale e biliare.
Sia la captazione dello ioduro che la sintesi della
tireoglobulina ed il rilascio degli ormoni tiroidei sono
controllati e stimolati dall’ormone tireotropo (Thyroid-Stimulating Hormone, TSH) la cui
increzione da parte delle cellule beta dell’adenoipofisi è,
a sua volta, stimolata dall’ormone ipotalamico TRH (Thyrotropin-Releasing Hormone; non
dosabile nel sangue periferico) ed inibita dalla iodotironine
circolanti, dalla dopamina e dalla somatostatina (vedi Par. 10.2). Dal punto di vista strutturale il
TSH è simile ad altri due ormoni glicoproteici ipofisari, l’ormone
follicolo stimolante (Follicle-Stimulating Hormone, FSH) e l’ormone
luteinizzante (Luteinizing Hormone, LH), e alla
gonadotropina corionica (human Chorionic Gonadotropin, hCG).
Tutti e quattro questi ormoni sono infatti degli eterodimeri
formati da una subunità
α non
specifica e da una subunità β diversa
per ciascuno di essi ![]() . Il TSH si lega a un
recettore specifico sulla superficie della
cellula follicolare determinando l’attivazione delle
proteine G modulatrici del sistema coinvolgente l’adenilato
cliclasi e le protein-chinasi A
. Il TSH si lega a un
recettore specifico sulla superficie della
cellula follicolare determinando l’attivazione delle
proteine G modulatrici del sistema coinvolgente l’adenilato
cliclasi e le protein-chinasi A
![]() . Un eccesso di
ioduro inibisce la risposta dell’adenilato ciclasi al TSH e
la iodinazione della tireoglobulina (effetto di Wolff-Chaikoff).
. Un eccesso di
ioduro inibisce la risposta dell’adenilato ciclasi al TSH e
la iodinazione della tireoglobulina (effetto di Wolff-Chaikoff).
L’ipotiroidismo viene distinto in primario e secondario. La causa più frequente di ipotiroidismo primario è una malattia autoimmune, la tiroidite di Hashimoto. Un’altra forma comune è l’ipotiroidismo iatrogeno che insorge in seguito ad una asportazione chirurgica della ghiandola o ad una terapia con iodio radioattivo. Anche l’uso di farmaci antitiroidei (propiltiouracile, metimazolo) può portare ad un ipotiroidismo che in genere recede dopo sospensione del trattamento. Un lieve ipotiroidismo si osserva frequentemente nelle donne anziane. Di più raro riscontro è attualmente il gozzo endemico che è dovuto a carenza di iodio nella alimentazione e che porta a grave deficienza mentale (cretinismo endemico). Altre rare cause sono i difetti enzimatici ereditari nella sintesi dell’ormone tiroideo che determinano la comparsa di ipotiroidismo associato a gozzo. L’ipotiroidismo secondario è dovuto ad una alterazione dell’asse ipotalamico-ipofisario per un deficit di increzione del TRH o del TSH.
Alcuni autori distinguono l’ipertiroidismo dalla tireotossicosi, intendendo con il primo termine (ipertiroidismo) le manifestazioni biochimiche e cliniche legate ad una iperproduzione ormonale da parte della tiroide e con il secondo termine (tireotossicosi) sia le manifestazioni precedenti sia quelle dovute ad una abnorme assunzione di farmaci a base di ormoni tiroidei o alla liberazione dell’ormone dalla ghiandola per fenomeni infiammatori distruttivi. Una classificazione schematica delle diverse forme di ipertiroidismo e tireotossicosi è riportata in Tab.10.I.
| Tab.10.I. Ipertiroidismo e tireotossicosi | |
| Aumentata captazione tiroidea di iodio in presenza di fattori plasmatici stimolanti la tiroide | |
| morbo di Basedow
|
|
| increzione inappropriata di TSH | |
| gravidanza molare
|
|
| coriocarcinoma | |
| iperemesi gravidica | |
| Aumentata captazione tiroidea di iodio in assenza di fattori plasmatici stimolanti la tiroide | |
| adenoma di Plummer
|
|
| ipertiroidismo non-autoimmune autosomico dominante | |
| Diminuita captazione tiroidea di iodio | |
| tiroiditi | |
| tireotossicosi fattizia
|
|
| ingestione di iodio | |
| cancro titoideo metastatico | |
| struma ovarii
|
|
Dal momento che le iodotironine circolano in gran parte legate
alle proteine, i loro livelli nel plasma dipendono anche dalla
concentrazione delle proteine leganti. Mutazioni genetiche
possono portare o ad un difetto o ad una iperproduzione della
globulina legante la tiroxina (TBG). Alcune di queste alterazioni
ereditarie sono
legate al cromosoma X, mentre altre, che
interessano probabilmente un gene regolatore, si trasmettono come carattere autosomico dominante. Un’altra
anomalia ereditaria è la
ipertiroxinemia eutiroidea familiare dovuta ad
una più elevata affinità dell’albumina o della prealbumina
per la T4. I soggetti con analbuminemia ereditaria (Par.
8.1.1) hanno
generalmente una maggiore concentrazione di TBG nel plasma. Un
aumento della TBG si può inoltre avere nel corso di gravidanza e
nell’iperestrogenismo, mentre un effetto opposto è prodotto
dagli androgeni. Alterazioni complesse nel livello degli ormoni
tiroidei e delle proteine plasmatiche che li veicolano sono di
frequente riscontro in malattie non-tiroidee (euthyroid sick syndrome). Un aumento
della frazione libera degli ormoni tiroidei può inoltre
osservarsi in pazienti trattati con eparina in quanto questa
sostanza stimola il rilascio di acidi grassi non esterificati che
competono con gli ormoni tiroidei per i siti leganti presenti
sull’albumina ![]() .
.
10.4.1. Metodi di determinazione
Nella maggior parte dei casi le concentrazioni plasmatiche di T3 e T4 variano in parallelo. Tuttavia nella tireotossicosi l’aumento della T3 totale è più spiccato, mentre nell’ipotiroidismo è la T4 totale a subire la diminuzione più marcata.
E’ stata attribuita particolare importanza alla determinazione della frazione degli ormoni tiroidei non legati alle proteine plasmatiche in quanto direttamente disponibili ad espletare la propria funzione biologica. L’interpretazione clinica del dato analitico non è tuttavia sempre facile, perché numerosi fattori possono far variare il livello delle iodotironine plasmatiche libere. D’altra parte, la determinazione degli ormoni liberi presenta delle difficoltà a causa sia della loro bassissima concentrazione (picomolare) rispetto a quella degli ormoni totali (nanomolare), sia della situazione di equilibrio reversibile fra la frazione libera e la frazione legata.
La determinazione del TSH è un importante test di
funzionamento tiroideo. Infatti, tranne in caso di ipotiroidismo
secondario a disfunzione ipofisaria, situazione piuttosto rara e
in ogni caso accompagnata da segni di deficit degli altri
ormoni ipofisari, un TSH normale indica che il paziente è
eutiroideo. Aumenti del TSH si riscontrano nell’ipotiroidismo
franco, nell’ipotiroidismo compensato o subclinico (quando l’aumentato
stimolo ipofisario determina la normalizzazione dei livelli di T4),
durante la fase di guarigione da malattie non tiroidee o in
occasione della ripresa dell’alimentazione dopo denutrizione.
Bassi livelli di TSH si ritrovano in pazienti con tireotossicosi
![]() ,
nell’ipertiroidismo subclinico (quando l’iperfunzione
della ghiandola non ha ancora provocato un’eccessiva
produzione di ormoni), dopo trattamento con dosi eccessive di
ormoni tiroidei, in corso di terapie con steroidi o con dopamina,
in corso di malattie sistemiche non tiroidee (soprattutto nell’anziano)
e nel digiuno prolungato.
,
nell’ipertiroidismo subclinico (quando l’iperfunzione
della ghiandola non ha ancora provocato un’eccessiva
produzione di ormoni), dopo trattamento con dosi eccessive di
ormoni tiroidei, in corso di terapie con steroidi o con dopamina,
in corso di malattie sistemiche non tiroidee (soprattutto nell’anziano)
e nel digiuno prolungato.
10.4.1a. Tiroxina
La tiroxina era un tempo stimata misurando la quantità di iodio presente in preparazioni parzialmente purificate di T4. Questi metodi comportavano la determinazione dello iodio in un precipitato proteico (Protein-Bound Iodine, PBI), nel suo estratto in butanolo (Butanol-Extractable Iodine, BEI) o in una frazione purificata mediante cromatografia su resina a scambio anionico. Tutte queste procedure, attualmente non più utilizzate, sono relativamente non-specifiche.
Un metodo più specifico e sensibile (Competitive Protein-Binding Assay, CPBA) si basa sulla competizione fra la T4 presente nel siero e quella radioattiva aggiunta al campione rispetto ad un numero limitato di siti ad alta affinità per l’ormone presenti su proteine. La radioattività legata alle proteine, che è misurata dopo aver rimosso la quota libera di T4 marcata, varia in funzione inversa rispetto alla concentrazione di T4 nel campione in esame. La T4 radioattiva è marcata con 125-I. La proteina adoperata nel saggio è una preparazione grezza di globulina legante la tiroxina (TBG) ottenuta per semplice diluizione del siero umano normale. Il legame fra la T4 e la prealbumina viene inibito selettivamente per aggiunta di tampone barbital, mentre la quota di T4 legata all’albumina è trascurabile nelle condizioni impiegate. La globulina legante la tiroxina presente nel campione in esame viene eliminata estraendo la T4 in etanolo prima del saggio.
Una sensibilità ed una specificità ancora più elevata possono essere ottenute sostituendo la globulina legante il retinolo con un anticorpo con una affinità verso la T4 fino a 10.000 volte superiore rispetto a quella della TBG. Questo metodo radioimmunologico (RadioImmunoAssay, RIA) non richiede una preventiva estrazione dell’analita dal campione in esame in quanto la globulina legante il retinolo presente in esso può essere inibita aggiungendo mertiolato, salicilato od acido 8-anilino-naftalensulfonico. La separazione della T4 radioattiva libera da quella legata può essere ottenuta usando anticorpi legati ad una matrice insolubile (vetro, plastica, cellulosa, particelle magnetiche), anticorpi anti-anticorpi o metodi di precipitazione in glicol polietilenico.
I dosaggi immunoenzimatici dell’ormone tiroideo utilizzano una T4 legata covalentemente ad un enzima e possono essere eseguiti in fase eterogenea od omogenea. Nel primo caso, viene utilizzato un anticorpo immobilizzato su un supporto solido (Enzyme-Linked ImmunoSorbent Assay, ELISA). Nel secondo caso (Enzyme-Multiplied Immunoassay Technique, EMIT) non è necessario separare il complesso antigene-anticorpo in quanto si sfrutta la proprietà dell’anticorpo di inibire l’attività enzimatica quando interagisce con la T4 coniugata alla lattico deidrogenasi. Entrambi i metodi necessitano un pretrattamento del siero in alcali per eliminare le interferenze.
Un ulteriore metodo, che non richiede un pretrattamento del
campione o una separazione delle fasi, utilizza un derivato
fosfonato della T4 che è un potente inibitore della
colinesterasi. Il legame dell’anticorpo alla T4 modificata
impedisce il legame dell’inibitore all’enzima, la cui
attività può essere direttamente dosata allo spettrofotometro
![]() .
.
I metodi immunofluorimetrici in fase omogenea (Polarization FluoroImmuno Assay, PFIA) si basano sulla misura della variazione della polarizzazione di fluorescenza della T4 marcata con fluoresceina in seguito al legame con l’anticorpo. Questi metodi richiedono un pretrattamento del campione in urea e dodecilsolfato per allontanare sostanze interferenti.
Una determinazione per diluizione isotopica si basa sull’aggiunta di una quantità nota di T4 deuterata al campione. L’ormone viene quindi purificato per estrazione in solvente organico, derivatizzato a N,O-bis(trifluoroacetil)-metilestere e sottoposto a gas-cromatografia associata a spettrometria di massa per determinare il rapporto fra le specie isotopiche presenti.
10.4.1b. Triiodotironina e tetraiodotirona libere
Le iodotironine libere nel siero possono essere separate da quelle legate alle proteine mediante tecniche di equilibrio di dialisi, ultrafiltrazione o cromatografia per gel filtrazione o per adsorbimento. Le iodotironine libere possono essere quindi dosate direttamente utilizzando anticorpi specifici, come già descritto nel Par. 10.4.1a. In alternativa, è possibile aggiungere direttamente il siero a degli anticorpi legati ad una matrice insolubile e, dopo il suo allontanamento, dosare con della T4 marcata i siti anticorpali ancora liberi: maggiore sarà la quota di radioattività adesa alla matrice insolubile, minore sarà la quantità di iodotironine libere nel campione in esame.
Tecniche indirette per la determinazione delle iodotironine libere prevedono l’aggiunta di un analogo radioattivo in tracce al campione in esame. La concentrazione delle iodotironine libere sarà desunta dalla quantità totale di iodotironine (determinata separatamente) e dal rapporto fra la radioattività libera e quella legata alle proteine. Per dosare quanto composto marcato non si lega alle proteine si può, ad esempio, aggiungere al campione una piccola quantità di T4 radioattiva, attendere che questa si equilibri con la T4 endogena (T4 binding), aggiungere un eccesso di anticorpi legati a delle perline di vetro e dosare la radioattività sul centrifugato dopo aver allontanato il plasma
Altre metodiche utilizzano degli anticorpi incapsulati in una matrice porosa e saturati con la T4 radioattiva. La matrice porosa funziona come una microcamera da dialisi, essendo permeabile all’ormone libero, ma non alle proteine. Quando la matrice è posta in contatto con il siero la T4 endogena si equilibra con quella radioattiva: la concentrazione delle iodotironine libere è così desunta dalla radioattività che rimane legata agli anticorpi incapsulati. La tecnica può essere ulteriormente migliorata sostituendo alla T4 radioattiva un suo analogo radioattivo che non è più in grado di legarsi alle proteine del siero, ma che conserva la capacità di legarsi all’anticorpo. In tal modo si evita di alterare gli equilibri fra ormoni liberi e legati nel siero.
Nessuno dei metodi proposti per il dosaggio delle iodotironine libere è del tutto soddisfacente. Non vi è accordo sulla temperatura, pH e forza ionica da usarsi durante le dialisi. Inoltre, una eccessiva diluizione del campione può portare a sovrastimare la frazione di T4 libera. Il materiale radioattivo aggiunto al campione nelle misure indirette può contenere contaminanti (generalmente iodio radioattivo derivante dalla degradazione dell’ormone stesso). Non si può inoltre escludere che il legame di questo materiale a delle proteine plasmatiche possa inficiare l’accuratezza della misura.
10.4.1c. Titolazione dei siti leganti sulle sieroproteine
I siti leganti liberi presenti sulle proteine del siero possono essere dosati utilizzando metodi radioattivi, fluorimetrici o enzimatici.
Quando si adoperano dei metodi radioattivi è preferibile usare della T3 marcata (T3 uptake) in quanto, avendo una minore affinità per la globulina legante la tiroxina (TBG), non sposta da questa proteina la T4 già presente nel campione in esame. Inoltre la T3 non si lega in quantità significative alla prealbumina e all’albumina. La T3 radioattiva in eccesso (che non ha reagito con la TBG) può essere misurata con vari mezzi. Il metodo più semplice consiste nell’aggiungere la T3 marcata al sangue intero e misurare la quota di T3 che, non legandosi alle proteine plasmatiche, penetra nei globuli rossi. Altre metodiche, più facilmente riproducibili, richiedono l’impiego di anticorpi legati ad una matrice solida, resine a scambio ionico, carbone attivo od altre sostanze adsorbenti.
Nel caso in cui si adoperi un metodo fluorimetrico diretto è necessario utilizzare la T4 legata alla fluoresceina, in quanto la T3 in tali condizioni perde la capacità di legarsi alle proteine plasmatiche. Il metodo consiste nel misurare la variazione di polarizzazione di fluorescenza del complesso fluoresceina-T4 aggiunto al siero in esame. E’ anche possibile adoperare della T3 marcata con fluoresceina, ma è necessario aggiungere in tal caso anche dell’anticorpo anti-T3 e della T3 non marcata capace di saturare tutti i siti disponibili presenti nel campione in esame. Si sfrutta infatti la capacità dell’anticorpo di legarsi al complesso fluoresceina-T3, quando non è impegnato nel legame con la T3 non marcata, determinando una diminuzione del segnale fluorescente.
Il metodo enzimatico si basa sull’uso del derivato
fosfonato della T4, un potente inibitore della colinesterasi.
Anche in questo caso non è possibile adoperare un derivato della
T3 perché perde la capacità di legarsi alle proteine
plasmatiche. La frazione libera del derivato della T4 è
calcolata dal grado di inibizione della colinesterasi
![]() .
.
10.4.1d. Ormone tireotropo ipofisario
La concentrazione dell’ormone tireotropo ipofisario (TSH)
è determinata mediante l’uso di anticorpi specifici
![]() .
Gli antisieri usati possono a volte mostrare una reattività
crociata con altri ormoni glicoproteici, in particolare con l’ormone
luteinizzante (LH) e la gonadotropina corionica (hCG)
.
Gli antisieri usati possono a volte mostrare una reattività
crociata con altri ormoni glicoproteici, in particolare con l’ormone
luteinizzante (LH) e la gonadotropina corionica (hCG)
![]() .
Le misure sono inoltre rese difficoltose per la disomogeneità
delle preparazioni di TSH usate come standard.
.
Le misure sono inoltre rese difficoltose per la disomogeneità
delle preparazioni di TSH usate come standard.
L’ormone può essere dosato mediante metodi
radioimmunologici (RIA) basati sulla competizione fra il TSH del
paziente ed una quantità nota di TSH radioattivo (aggiunto
al campione) rispetto ad un numero limitato di siti anticorpali.
Metodi di tipo non-competitivo consistono invece nel dosare il
TSH mediante due diversi anticorpi, aggiunti in eccesso e diretti contro
differenti siti antigenici in modo da formare un complesso a "sandwich":
uno dei due anticorpi è legato ad una matrice insolubile in modo da permettere
una facile separazione del prodotto di reazione, mentre l’altro è reso
radioattivo per aggiunta di 125-I (metodo IRMA) o è coniugato ad un enzima
(generalmente a della fosfatasi alcalina; metodo ELISA) per permettere un
dosaggio quantitativo del complesso antigene-anticorpo. Un altro metodo ancora
utilizza un anticorpo anti-TSH insolubilizzato, un
anticorpo anti-TSH legato alla biotina e una perossidasi da
rafano legata all’avidina, la cui attività è dosabile per
aggiunta di un cromogeno. Si ha così il vantaggio di legare una
molecola più piccola ![]() all’anticorpo solubile che dovrà
contribuire alla formazione del complesso a "sandwich",
evitando l’insorgenza di impedimenti sterici nel legame.
all’anticorpo solubile che dovrà
contribuire alla formazione del complesso a "sandwich",
evitando l’insorgenza di impedimenti sterici nel legame.
Altre tecniche prevedono l’uso di un tracciante fluorescente o luminescente
![]() . Un ostacolo per una determinazione fluorimetrica del complesso macromolecolare insolubilizzato è
rappresentato dall’elevata fluorescenza di base dei
campioni di siero, che non può essere ovviamente eliminata, nel
caso di una determinazione del TSH, precipitando le proteine. Una
soluzione è quella di marcare l’anticorpo con l’europio,
la cui fluorescenza ha una emivita nell’ordine dei
microsecondi (l’emivita della fluorescenza del siero è nell’ordine
dei nanosecondi), e di eseguire misure di fluorescenza risolte
nel tempo (Time-Resolved ImmunoFluoroMetric Assay, TR-IFMA).
. Un ostacolo per una determinazione fluorimetrica del complesso macromolecolare insolubilizzato è
rappresentato dall’elevata fluorescenza di base dei
campioni di siero, che non può essere ovviamente eliminata, nel
caso di una determinazione del TSH, precipitando le proteine. Una
soluzione è quella di marcare l’anticorpo con l’europio,
la cui fluorescenza ha una emivita nell’ordine dei
microsecondi (l’emivita della fluorescenza del siero è nell’ordine
dei nanosecondi), e di eseguire misure di fluorescenza risolte
nel tempo (Time-Resolved ImmunoFluoroMetric Assay, TR-IFMA).
10.4.2. Preparazione del campione ed intervalli di riferimento
Il siero è generalmente preferito al plasma nel dosaggio degli ormoni tiroidei. I campioni per la determinazione delle iodotironine libere non dovrebbero contenere eparina. Dovrebbero essere inoltre esclusi i campioni emolizzati o lipemici. Ripetuti congelamenti e scongelamenti del siero possono portare ad una denaturazione dell’ormone tireotropo e/o delle proteine leganti le iodotironine.
Il silicone presente nelle provette può alterare alcuni dosaggi immunologici in fase solida. Interferenze sui dosaggi immunologici della T4 possono derivare inoltre dalla presenza di autoanticorpi che è possibile riscontrare sia in soggetti eutiroidei, sia in pazienti con malattie tiroidee o del sistema immunitario (macroglobulinemia di Waldenström).
A causa della scarsa accuratezza di molte delle determinazioni, è consigliabile che ciascun laboratorio determini autonomamente i propri intervalli di riferimento per le analisi riguardanti le patologie tiroidee. I valori di riferimento generalmente accettati per la T4 nel siero sono 45-115 μg/L (58-148 nmol/L), mentre nei bambini i valori sono leggermente più alti (62-170 μg/L). In condizioni di equilibrio, le concentrazioni di T4 e T3 libere sono rispettivamente circa 32 ng/L e 4 ng/L. I valori di riferimento per il TSH sono 0,51-5,75 mU/L; nel neonato la concentrazione del TSH nel siero aumenta di 10-20 volte subito dopo la nascita, per tornare ai livelli dell’adulto dopo circa 5 giorni. Dopo stimolazione con l’ormone ipotalamico TRH, il livello del TSH aumenta di 2-20 mU/L. Aumenti minori sono osservati in pazienti con ipotiroidismo secondario (ipofisario), mentre variazioni più marcate sono riscontrabili in soggetti affetti da ipotiroidismo primario (tiroideo). Nel caso di ipotiroidismo terziario (ipotalamico) la risposta al TRH è di ampiezza normale ma significativamente ritardata.
L’incorporazione della T3 (T3 uptake, Par. 10.4.1c) viene valutata come rapporto fra la radioattività adsorbita dal siero del paziente e quella adsorbita da un siero normale di controllo; i valori di riferimento sono 0,83-1,17. Uno dei parametri più comunemente richiesto è l’indice di T4 libera (free thyroxine index, FTI) che è il prodotto fra il suddetto rapporto (T3 uptake ratio) e la concentrazione totale della T4 nel siero. L’intervallo di riferimento per l’FTI è 37,4-134 μg/L.
10.5. ORMONI CORTICOSURRENALI
Istologicamente la corteccia surrenale consta di tre zone in diretta continuità tra di loro: la zona glomerulare, la zona fascicolata e la zona reticolare. La prima zona è deputata alla sintesi dei mineralcorticoidi, mentre le altre due sono deputate alla sintesi degli ormoni glicoattivi ed androgeni. La zona glomerulare, a ridosso della capsula, è la più esterna e sottile ed è formata da gruppi di piccole cellule relativamente povere di inclusioni lipidiche. La zona fascicolata si distingue per la disposizione colonnare delle travate cellulari che, almeno nel loro terzo esterno, sono costituite da spongiociti notevolmente carichi di lipidi. La zona reticolare è caratterizzata da travate disposte a reticolo, costituite da cellule contenenti particolari inclusioni (corpi siderofili, pigmento bruno); questa zona si continua con la midollare del surrene.
Gli ormoni della corteccia surrenale sono degli steroidi contenenti lo scheletro carbonioso del ciclopentanoperidrofenantrene, proprio del colesterolo (vedi Par. 3.3). L’attività biologica è condizionata da modificazioni chimiche su questa struttura di base: i composti a 21 atomi di carbonio hanno attività progestinica, glucocorticoide e mineralcorticoide, mentre la rottura del legame fra C-17 e C-20 porta alla formazione di steroidi a 19 atomi di carbonio con attività androgenica.
La conversione del colesterolo in corticosteroidi richiede l’intervento di una serie di enzimi in parte mitocondriali e in parte microsomiali. Ad eccezione della 3b-idrossisteroide deidrogenasi (NAD-dipendente), tutti questi enzimi sono delle ossidasi a funzione mista appartenenti alla famiglia dei citocromi P450. Le ossidasi mitocondriali richiedono la presenza dell’adrenodossina e dell’adrenodossina reduttasi, mentre le ossidasi microsomiali utilizzano la citocromo P450 reduttasi. Entrambi i sistemi accettano elettroni dall’NADPH (Fig. 10.6)
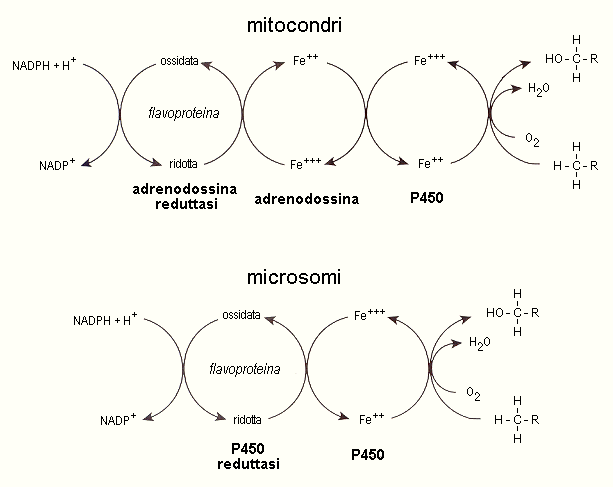
Fig. 10.6. Ossidasi a funzione mista coinvolte nella steroidogenesi. Trasporto degli elettroni dall’NADPH al citocromo P450 nei mitocondri e nei microsomi.
Uno schema delle reazioni coinvolte nella sintesi
degli ormoni steroidei è mostrato in Fig. 10.7.
Gli enzimi che catalizzano queste reazioni sono variamente
espressi nelle cellule in cui è attiva la steroidogenesi. In tutte queste
cellule sono presenti l’enzima di scissione della
catena laterale del colesterolo (desmolasi) e la 3β-idrossisteroide
deidrogenasi / Δ5-4 isomerasi
![]() . Il primo enzima opera la conversione del
colesterolo in pregnenolone a livello mitocondriale, catalizzando
tre reazioni consecutive: l’idrossilazione in posizione 20α, l’idrossilazione in posizione 22 e
la scissione del legame fra C-20 e C-22. La 3β-idrossisteroide
deidrogenasi / Δ5-4 isomerasi, presente nei microsomi, opera la conversione del
pregnenolone in progesterone, del 17-idrossipregnenolone in 17-idrossiprogesterone
e del deidroepiandrosterone in androstenedione.
. Il primo enzima opera la conversione del
colesterolo in pregnenolone a livello mitocondriale, catalizzando
tre reazioni consecutive: l’idrossilazione in posizione 20α, l’idrossilazione in posizione 22 e
la scissione del legame fra C-20 e C-22. La 3β-idrossisteroide
deidrogenasi / Δ5-4 isomerasi, presente nei microsomi, opera la conversione del
pregnenolone in progesterone, del 17-idrossipregnenolone in 17-idrossiprogesterone
e del deidroepiandrosterone in androstenedione.
La conversione del pregnenolone in 17-idrossipregnenolone
e del progesterone in 17-idrossiprogesterone, che porta alla
formazione di ormoni glicoattivi ed androgeni, è catalizzata
da un enzima microsomiale bifunzionale (17α-idrossilasi
/ 17-20 liasi) che
è presente nelle zone interne della corteccia surrenale e nelle
gonadi, ma è assente nella zona glomerulare dei surreni
![]() . La 21-idrossilasi microsomiale, necessaria per la sintesi degli ormoni glicoattivi
e mineralcorticoidi, è invece presente nelle zone sia interne
che esterne della corteccia surrenale.
Le tappe finali della steroidogenesi corticosurrenalica sono catalizzate da due
enzimi mitocondriali molto simili fra loro. Il primo, presente
nella zona glomerulare dei surreni, catalizza l’idrossilazione
dell’11-deossicorticosterone in posizione 11β e 18, nonché l’ossidazione in
posizione 18, e porta alla formazione dell’aldosterone. Il
secondo enzima, presente nelle zone interne della corticale
surrenale, catalizza esclusivamente l’idrossilazione in
posizione 11β, convertendo l’11-deossicortisolo
in cortisolo.
. La 21-idrossilasi microsomiale, necessaria per la sintesi degli ormoni glicoattivi
e mineralcorticoidi, è invece presente nelle zone sia interne
che esterne della corteccia surrenale.
Le tappe finali della steroidogenesi corticosurrenalica sono catalizzate da due
enzimi mitocondriali molto simili fra loro. Il primo, presente
nella zona glomerulare dei surreni, catalizza l’idrossilazione
dell’11-deossicorticosterone in posizione 11β e 18, nonché l’ossidazione in
posizione 18, e porta alla formazione dell’aldosterone. Il
secondo enzima, presente nelle zone interne della corticale
surrenale, catalizza esclusivamente l’idrossilazione in
posizione 11β, convertendo l’11-deossicortisolo
in cortisolo.
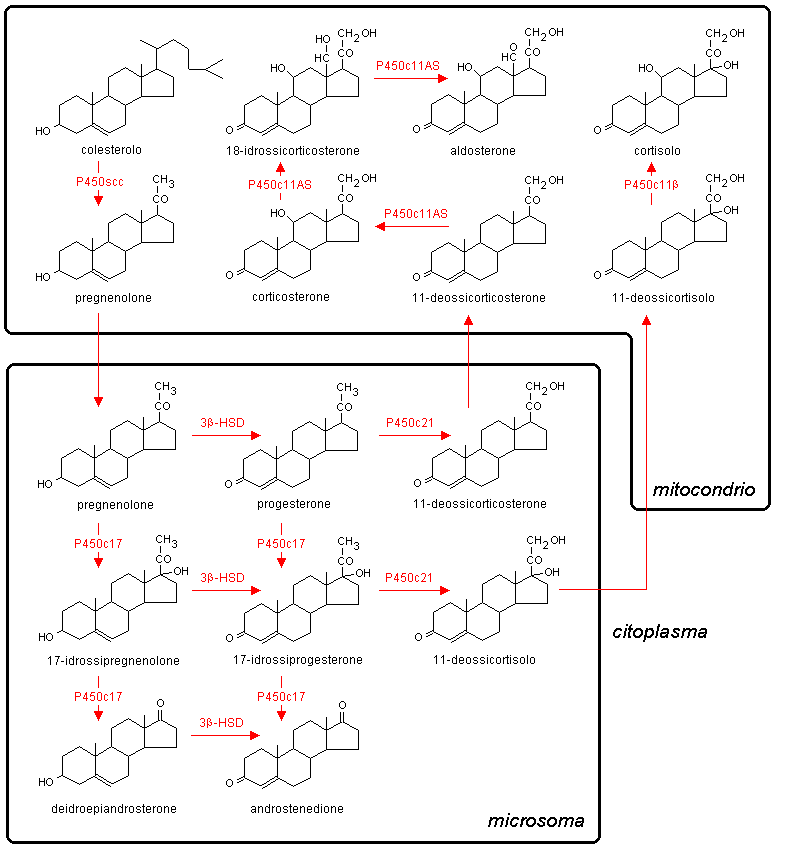
Fig. 10.7. Biosintesi degli steroidi corticosurrenali. La figura riunisce in un unico schema reazioni che in realtà avvengono in zone della corteccia surrenale anatomicamente e funzionalmente distinte, come è indicato nel testo. P450scc: enzima di scissione della catena laterale del colesterolo; P450c17: 17α-idrossilasi / 17,20 liasi; P450c21: 21-idrossilasi; P450c11β: 11β-idrossilasi; P450c11AS: aldosterone sintetasi; 3β-HSD: 3β-idrossisteroide deidrogenasi / Δ5-4 isomerasi.
Gli ormoni steroidei circolano in gran parte legati alle
proteine plasmatiche. Il cortisolo, principale glucocorticoide
ematico, è presente in circolo in forma libera solo in una
piccola quota (circa l’8%), per la restante parte è
veicolato dalla
transcortina (CBG, Corticosteroid-Binding
Globulin) e in minor misura dall’albumina. Anche il
progesterone ha una elevata affinità per la transcortina e, nell’ultimo
periodo della gravidanza, può competere con il cortisolo per il
legame a questa proteina di trasporto
![]() . Il corticosterone
si lega alla transcortina come il cortisolo, mentre l’aldosterone
(principale mineralcorticoide ematico presente per il 30-40% in
forma libera) ha una ridotta affinità per questa proteina. L’androstenedione
e il deidroepiandrosterone circolanti si legano in maggior misura
alla globulina legante gli ormoni sessuali (SHBG, Sex
Hormone-Binding Globulin), ma con una affinità minore
rispetto al testosterone ed al diidrotestosterone (vedi Par. 10.6). Dal momento
che la forma attiva degli ormoni steroidei è quella libera, il
legame con le proteine plasmatiche permette di costituire una
riserva di ormoni circolanti capace di tamponare le marcate
oscillazioni della concentrazione delle forme libere.
. Il corticosterone
si lega alla transcortina come il cortisolo, mentre l’aldosterone
(principale mineralcorticoide ematico presente per il 30-40% in
forma libera) ha una ridotta affinità per questa proteina. L’androstenedione
e il deidroepiandrosterone circolanti si legano in maggior misura
alla globulina legante gli ormoni sessuali (SHBG, Sex
Hormone-Binding Globulin), ma con una affinità minore
rispetto al testosterone ed al diidrotestosterone (vedi Par. 10.6). Dal momento
che la forma attiva degli ormoni steroidei è quella libera, il
legame con le proteine plasmatiche permette di costituire una
riserva di ormoni circolanti capace di tamponare le marcate
oscillazioni della concentrazione delle forme libere.
Gli steroidi sono catabolizzati principalmente a livello
epatico. Il cortisolo viene inattivato a cortisone dalla 11β-idrossisteroide deidrogenasi
![]() . Altri cataboliti sono il tetraidrocortisolo, il
tetraidrocortisone e il tetraidroaldosterone. Questi composti
vengono quindi glucuronati ed escreti con le urine e, in minor
misura, con la bile.
. Altri cataboliti sono il tetraidrocortisolo, il
tetraidrocortisone e il tetraidroaldosterone. Questi composti
vengono quindi glucuronati ed escreti con le urine e, in minor
misura, con la bile.
Gli androgeni sono catabolizzati in parte dal fegato, ma in
misura maggiore a livello delle gonadi, della pelle e del tessuto
adiposo. Nel fegato, nei surreni e nelle gonadi il
deidroepiandrosterone è esterificato dall’acido solforico
in posizione 3β; la reazione è
catalizzata dalla solfotransferasi che utilizza la 3’-fosfoadenosina 5’-fosfosolfato ![]() come dotanore di
solfato. Il deidroepiandrosterone solfato è un precursore degli
estrogeni prodotti dalla placenta; il composto viene prima
desolfatato e poi trasformato in estrone dall’aromatasi.
Androstenedione è trasformato in testosterone dalla 17β-idrossisteroide
deidrogenasi
come dotanore di
solfato. Il deidroepiandrosterone solfato è un precursore degli
estrogeni prodotti dalla placenta; il composto viene prima
desolfatato e poi trasformato in estrone dall’aromatasi.
Androstenedione è trasformato in testosterone dalla 17β-idrossisteroide
deidrogenasi
![]() .
.
10.5.1. Controllo della increzione ormonale
La corteccia surrenale può rispondere ai meccanismi di controllo in modo acuto o cronico. La risposta acuta si manifesta dopo pochi minuti ed è da mettere in relazione con un aumento della disponibilità di colesterolo, utilizzato nella tappa iniziale del processo biosintetico. L’incorporazione di colesterolo nel mitocondrio è indotta dal cAMP ed è controllata da una fosfoproteina mitocondriale denominata StAR (Steroidogenic Acute Regulatory Protein). La fase cronica della risposta della corteccia surrenale richiede diverse ore o giorni ed è dovuta ad un aumento della trascrizione degli enzimi coinvolti nella steroidogenesi.
10.5.1a. Asse ipotalamo-ipofisi-surrene
L’attività dell’enzima di scissione della catena
laterale del colesterolo, che limita cineticamente tutta la
steroidogenesi, è stimolata dalla corticotropina ipofisaria (ACTH,
Adrenocorticotropic Hormone). Questo ormone è un
peptide di 39 aminoacidi, che deriva da una frammentazione
della pro-opiomelanocortina sintetizzata dalle cellule del
sistema adenoipofisario ![]() . L’increzione di ACTH
è, a sua volta, stimolata in maniera sinergica dalla vasopressina (vedi
Par. 10.1) e dall’ormone rilasciante la corticotropina (CRH, Corticotropin-Releasing Hormone),
un peptide di 41 aminoacidi derivante dalla proteolisi di un
preproormone di 191 aminoacidi, che è formato a livello dell’eminenza
mediana dell’ipotalamo ed è versato nel sistema portale
ipofisario. Il CRH è presente normalmente in concentrazioni
bassissime nel sangue periferico, ma aumenta in questo settore
durante la gravidanza in quanto viene prodotto dalla placenta. Nel sangue periferico della donna gravida il CRH
raggiunge livelli simili a quelli del sistema portale ipofisario
e si lega ad una specifica proteina di trasporto (CRH-binding protein) che lo disattiva (vedi
Par. 10.7.1).
La increzione ipotalamica del CRH è inibita dai glicocorticoidi
ed è modulata in vario modo da diversi fattori: steroidi
sessuali, agenti adrenergici, citochine (interleuchina 1 e 6,
fattore di necrosi tumorale α), stress
. L’increzione di ACTH
è, a sua volta, stimolata in maniera sinergica dalla vasopressina (vedi
Par. 10.1) e dall’ormone rilasciante la corticotropina (CRH, Corticotropin-Releasing Hormone),
un peptide di 41 aminoacidi derivante dalla proteolisi di un
preproormone di 191 aminoacidi, che è formato a livello dell’eminenza
mediana dell’ipotalamo ed è versato nel sistema portale
ipofisario. Il CRH è presente normalmente in concentrazioni
bassissime nel sangue periferico, ma aumenta in questo settore
durante la gravidanza in quanto viene prodotto dalla placenta. Nel sangue periferico della donna gravida il CRH
raggiunge livelli simili a quelli del sistema portale ipofisario
e si lega ad una specifica proteina di trasporto (CRH-binding protein) che lo disattiva (vedi
Par. 10.7.1).
La increzione ipotalamica del CRH è inibita dai glicocorticoidi
ed è modulata in vario modo da diversi fattori: steroidi
sessuali, agenti adrenergici, citochine (interleuchina 1 e 6,
fattore di necrosi tumorale α), stress
![]() .
L’asse CRH-ACTH, oltre a stimolare la steroidogenesi
surrenalica, è coinvolto anche nel controllo del comportamento
alimentare attraverso un’interazione con il sistema della
leptina.
.
L’asse CRH-ACTH, oltre a stimolare la steroidogenesi
surrenalica, è coinvolto anche nel controllo del comportamento
alimentare attraverso un’interazione con il sistema della
leptina.
10.5.1b. Sistema renina-angiotensina
La increzione dell’aldosterone è regolata principalmente dal sistema renina-angiotensina. La renina è un enzima sintetizzato dalle cellule iuxtaglomerulari del rene (vedi Par. 11) che agisce su di un substrato costituito da una α2-globulina (angiotensinogeno) dando luogo alla formazione di un decapeptide (angiotensina I). L’angiotensina I è, a sua volta, attivata nel sangue e nei tessuti da un enzima convertente che forma l’octapeptide angiotensina II. Questa, come anche l’angiotensina III, che deriva dall’angiotensina II per rimozione dell’aspartato N-terminale, stimola la sintesi dell’aldosterone e contemporaneamente aumenta le resistenze arteriose periferiche determinando un aumento della pressione arteriosa. La trasformazione dell’angiotensina I in angiotensina II avviene in gran parte durante il primo passaggio della sostanza attraverso il polmone. L’angiotensina II è rapidamente inattivata nei tessuti e nel plasma dalle peptidasi. Le angiotensine II e III agiscono sulle cellule della zona glomerulare del surrene legandosi a recettori di membrana con conseguente potenziamento dell’attività dell’enzima di scissione della catena laterale del colesterolo e dell’aldosterone sintetasi. Il rilascio di renina dalle cellule iuxtaglomerulari è regolato dai barocettori renali, dal sistema nervoso adrenergico e dal livello di sodiemia: un aumento dell’increzione di aldosterone stimola la ritenzione renale di sodio e di acqua, determina un aumento della pressione sanguigna, provoca uno stiramento delle arteriole renali e diminuisce infine la produzione di renina. L’iperpotassiemia stimola direttamente la produzione di aldosterone, mentre l’ipopotassiemia diminuisce la risposta delle cellule della zona glomerulare del surrene all’angiotensina II.
10.5.1c. Regolazione della increzione degli androgeni surrenali
La increzione degli ormoni glicoattivi e degli androgeni non
è strettamente accoppiata. La concentrazione degli androgeni nel
bambino è estremamente bassa ed aumenta notevolmente alla pubertà
senza alcuna modificazione nel livello di ACTH e di cortisolo
![]() . La
concentrazione degli androgeni diminuisce nuovamente dopo i 30-40
anni, ma anche in questo caso non si osserva alcuna variazione
nella produzione dei glicocorticoidi
. La
concentrazione degli androgeni diminuisce nuovamente dopo i 30-40
anni, ma anche in questo caso non si osserva alcuna variazione
nella produzione dei glicocorticoidi
![]() .
.
10.5.1d. Malattie legate ad una disfunzione corticosurrenale
Un aumento in circolo di ormoni glicocorticoidi di origine corticosurrenale o esogena caratterizza la sindrome di Cushing. Questa può essere dovuta nel 60-70% dei casi ad una iperfunzione corticosurrenale conseguente ad una eccessiva produzione di ACTH ipofisario (malattia di Cushing), nel 15-20% dei casi ad un tumore extraipofisario secernente ACTH (carcinoma polmonare, carcinoma tiroideo, timoma, feocromocitoma, carcinoide bronchiale o di altre sedi, tumore delle insule pancreatiche) e nel 8-10% dei casi ad una iperproduzione di glicocorticoidi ACTH-indipendente (conseguente ad un adenoma o ad un carcinoma surrenalico).
L’iperaldosteronismo può essere primario o secondario. La causa dell’iperaldosteronismo primario può essere rappresentata da un adenoma o da una iperplasia bilaterale della zona glomerulare del surrene. L’iperaldosteronismo secondario può derivare da una stimolazione del sistema renina-angiotensina per un tumore secernente renina, una riduzione del volume ematico o un abuso di lassativi e diuretici. Una classificazione delle ipernatriemie ipopatassiemiche da eccesso di ormoni surrenalici è riportata in Tab. 12.III.
L’ipofunzione corticosurrenale può essere imputata a fattori intrinseci al surrene, a difetti di increzione di ACTH o ad una mancata risposta periferica all’ormone. Una classificazione schematica delle diverse forme di ipofunzione corticosurrenale è riportata in Tab. 10.II.
10.5.2. Metodi di determinazione
Le indagini biochimiche sulla funzionalità della corteccia surrenale riguardano il dosaggio degli steroidi e degli ormoni peptidici correlati a questa ghiandola. Alcuni metodi utilizzano dei reagenti chimici relativamente aspecifici capaci interagire con intere classi di composti, ma non di discriminare all’interno di queste classi. Altri metodi (generalmente immunologici) hanno invece una elevata specificità.
10.5.2a. Indagini colorimetriche aspecifiche
I 17-idrossicorticosteroidi urinari comprendono quei composti
evidenziabili con la reazione di Porter-Silber (Fig. 10.8). Fanno parte di questo gruppo i
principali cataboliti urinari del cortisolo (tetraidrocortisolo e
tetraidrocortisone) e l’11-desossicortisolo. La possibilità
di dosare simultaneamente i derivati sia del cortisolo che dell’11-desossicortisolo
è utile per valutare la risposta al metirapone, un inibitore
dell’11-idrossilasi. L’assunzione di numerosi farmaci (tra
cui lo spironolattone ![]() e il
clordiazepossido
e il
clordiazepossido ![]() ) tuttavia determina un falso aumento dei
valori osservati. Il metodo prevede l’idrolisi dei
glucuronoderivati degli steroidi e la loro estrazione in
cloroformio e diclorometano. L’estratto viene lavato in
alcali e fatto reagire con la fenilidrazina a caldo.
) tuttavia determina un falso aumento dei
valori osservati. Il metodo prevede l’idrolisi dei
glucuronoderivati degli steroidi e la loro estrazione in
cloroformio e diclorometano. L’estratto viene lavato in
alcali e fatto reagire con la fenilidrazina a caldo.
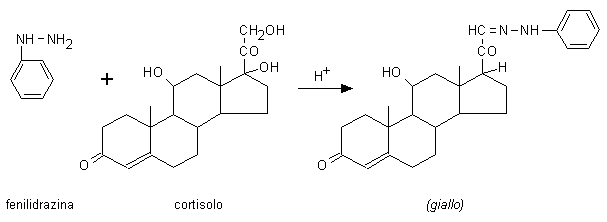
Fig. 10.8. Reazione di Porter-Silber
Sono denominati 17-chetosteroidi urinari quegli steroidi che prendono parte alla reazione di Zimmermann (Fig.10.9). Essi sono rappresentati principalmente dal 5α-androsterone e dal 5β-androsterone (etiocolanolone), che si trovano nelle urine in gran parte come solfati. Il loro principale precursore ematico è il deidroepiandrosterone, ma anche altri steroidi (testosterone, androstenedione, cortisolo, etc.) contribuiscono alla loro formazione. La determinazione dei 17-chetosteroidi indaga perciò la funzionalità sia del surrene che delle gonadi: nella donna circa il 90% dei 17-chetosteroidi è di origine surrenalica, mentre nell’uomo questa quota scende al 60-70%. Per eseguire la reazione di Zimmermann, i chetosteroidi coniugati devono essere prima idrolizzati in acido a caldo ed estratti in solvente organico (benzene, metilencloruro, etilendicloruro). Dopo aver rimosso i fenoli ed altri pigmenti interferenti con soda, i chetosteroidi vengono fatti reagire con il m-dinitrobenzene che forma un addotto di colore violetto. In alternativa, la reazione può essere eseguita in un mezzo acquoso in presenza di un sale ammonico quaternario che solubilizza sia gli steroidi che il m-dinitrobenzene.
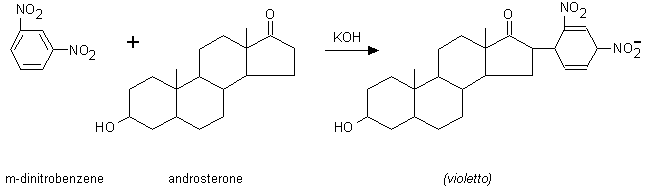
Fig. 10.5. Reazione di Zimmermann
Gli steroidi 17-chetogenici urinari sono composti che possono essere convertiti in 17-chetosteroidi mediante ossidazione con bismutato di sodio. Fanno parte di questo categoria gli steroidi con in posizione 17 un gruppo laterale formato dal diidrossiacetone (cortisone, cortisolo, 11-desossicortisolo e loro derivati), dal glicerolo (cortolo e cortolone, derivanti dal cortisolo e dal cortisone per riduzione del gruppo chetonico in posizione 20) o dal 17-20-glicole (pregnantriolo). Il dosaggio dei 17-chetogenici urinari è attualmente obsoleto per l’eterogeneità dei composti appartenenti a questo gruppo.
Il cortisolo può essere convertito in un composto fluorescente in presenza di etanolo ed acido solforico anidro. Numerose sostanze interferenti sono preventivamente eliminate estraendo lo steroide in solvente organico (diclorometano).
10.5.2b. Metodi specifici
Metodi altamente specifici per il dosaggio dei corticosteroidi e degli ormoni peptidici correlati al surrene utilizzano tecniche immunologiche o di cromatografia liquida ad alta pressione, gas cromatografia e spettrometria di massa.
I metodi immunologici sono generalmente basati
sulla competizione, rispetto ad un numero limitato di siti
anticorpali, del campione in esame con una preparazione standard
marcata con un isotopo radioattivo, con un fluoroforo o con un
enzima. L’analisi può essere eseguita in fase eterogenea (RIA)
od omogenea (FPIA, EMIT
![]() ).
Le due ultime metodiche sono più rapide e facilmente
automatizzabili. Nei sistemi EMIT, gli enzimi usati come
marcatori possono risentire anche fortemente di variazioni di pH
e temperatura e della presenza di adulteranti.
).
Le due ultime metodiche sono più rapide e facilmente
automatizzabili. Nei sistemi EMIT, gli enzimi usati come
marcatori possono risentire anche fortemente di variazioni di pH
e temperatura e della presenza di adulteranti.
Nella valutazione del sistema renina-angiotensina è di gran lunga più diffuso il dosaggio dell’attività reninica plasmatica che consiste nel determinare la quantità di angiotensina I che si forma per unità di tempo incubando il siero del paziente in condizioni standard. L’attività enzimatica può dipendere dalla quantità di angiotensinogeno presente nel siero in esame, specie in pazienti ipo-disprotidemici.
10.5.3. Preparazione del campione ed intervalli di riferimento
Per il dosaggio dei corticosteroidi ematici si possono usare indifferentemente campioni di siero o di plasma. Gli steroidi sono spesso determinati in campioni di urine delle 24 ore. In tal caso, per una migliore conservazione dell’analita, è consigliabile raccogliere le urine in un contenitore in cui è stato aggiunto dell’acido borico (10 g/L) o dell’acido acetico (7 mL/L).
L’ACTH è una molecola estremamente labile che si deteriora rapidamente se il campione di plasma non è rapidamente separato dalla parte corpuscolata. Si consiglia di raccogliere il sangue in provette di plastica contenenti EDTA o eparina, di trasportare il campione rapidamente in ghiaccio nel laboratorio e di centrifugare nuovamente il plasma prima di conservarlo a -70° C.
La concentrazione del cortisolo ematico presenta una variazione circandiana con un minimo intorno alla mezzanotte ed un massimo alle 8-9 del mattino. Al fine di valutare correttamente i valori dell’ormone circolante, è necessario eseguire più prelievi nel corso della giornata (solitamente alle ore 8, 16 e 24). I valori comunemente osservati sono compresi tra 5 e 25 μg/dL nel campione del mattino, con una riduzione di almeno il 50% nel campione della sera. Un aumento della concentrazione ematica di cortisolo viene osservato in gravidanza o dopo l’uso di contraccettivi orali. L’escrezione urinaria nelle 24 ore è normalmente inferiore a 150 μg.
Il dosaggio dell’aldosterone presenta maggiori difficoltà a causa dei suoi bassissimi livelli che, oltre a presentare una ritmicità circandiana, tendono ad aumentare con l’ortostatismo o a seguito di un incremento del potassio o una diminuzione del sodio nella dieta. La concentrazione ematica nelle prime ore del mattino è di 30-100 pg/mL in clinostatismo e di 50-300 pg/mL in ortostatismo. L’escrezione urinaria normale giornaliera di aldosterone in soggetti che assumano circa 120 mEq di sodio al giorno è di 5-20 μg.
In condizioni normali, l’escrezione urinaria di 17-idrossicorticosteroidi nelle 24 ore è di 5-15 mg nei maschi e lievemente inferiore nelle femmine. L’escrezione di 17-chetosteroidi è di 8-20 mg nei maschi e 6-15 mg nelle femmine, mentre quella degli steroidi 17-chetogenici è di 6-20 mg.
Il livello ematico di ACTH varia parallelamente con il livello di cortisolo, tuttavia l’interpretazione del dato analitico è difficile a causa dell’andamento pulsatile dell’increzione ormonale. I valori sono di 25-100 pg/mL al mattino ed inferiori a 50 pg/mL di sera. Gli stati di stress, la gravidanza ed il ciclo mestruale determinano un aumento dei valori di ACTH fino a 600 pg/mL. La determinazione dei livelli plasmatici di ACTH è utile per differenziare l’insufficienza surrenalica primaria da quella secondaria. Nel primo caso si osservano bassi valori di cortisolo ed alti valori di ACTH (> 200 pg/mL), mentre nelle forme secondarie la concentrazione di ACTH è nei limiti della norma o diminuita (< 50 pg/mL).
L’attività reninica plasmatica è generalmente di 0,5-3,0 ng/(mL x h) in clinostatismo e di 2-7 ng/(mL x h) in ortostatismo in soggetti con un normale contenuto di sodio e potassio nella dieta.
10.6. ORMONI DELLE GONADI FEMMINILI
L’ovario è costituito da una zona midollare e da una zona corticale più
esterna, comprendente uno stroma connettivale, che dà luogo ad una specie di
capsula fibrosa alla periferia dell’organo, ed un elevato numero di follicoli. Ciascun follicolo
è formato da una cellula germinale (oocita) circondata da una membrana
propria (zona pellucida) e da uno strato di cellule epiteliali (granulosa). Le cellule della granulosa non sono a diretto contatto con il sangue in
quanto sono separate, per mezzo della lamina basale, dalla teca che è,
invece, vascolarizzata
![]() . Le cellule della granulosa sono connesse fra loro e con
l’oocita (attraverso prolungamenti citoplasmatici che attraversano la zona
pellucida) per mezzo di giunzioni che permettono un interscambio di metaboliti.
. Le cellule della granulosa sono connesse fra loro e con
l’oocita (attraverso prolungamenti citoplasmatici che attraversano la zona
pellucida) per mezzo di giunzioni che permettono un interscambio di metaboliti.
Il principale ormone steroideo prodotto dalle cellule della granulosa è l’estradiolo. Per la sintesi di questo ormone è necessaria una
interconnessione metabolica fra le cellule della granulosa e quelle adiacenti della teca.
Infatti, questo ultime cellule producono il deidroepiandrosterone e l’androstenedione
che, trasferito nella granulosa, diviene substrato per la sintesi degli
estrogeni (vedi Fig. 10.10). L’androstenedione
è sintetizzato nelle cellule della teca a partire dal colesterolo attraverso l’azione combinata
dell’enzima di scissione della catena laterale del colesterolo, della 17α-idrossilasi
/ 17-20 liasi
![]() e
della 3β-idrossisteroide
deidrogenasi / Δ5-4 isomerasi
e
della 3β-idrossisteroide
deidrogenasi / Δ5-4 isomerasi
![]() . Gli
enzimi presenti nella granulosa, che intervengono nella sintesi dell’estradiolo,
sono l’aromatasi e la 17b-idrossisteroide
deidrogenasi.
. Gli
enzimi presenti nella granulosa, che intervengono nella sintesi dell’estradiolo,
sono l’aromatasi e la 17b-idrossisteroide
deidrogenasi.
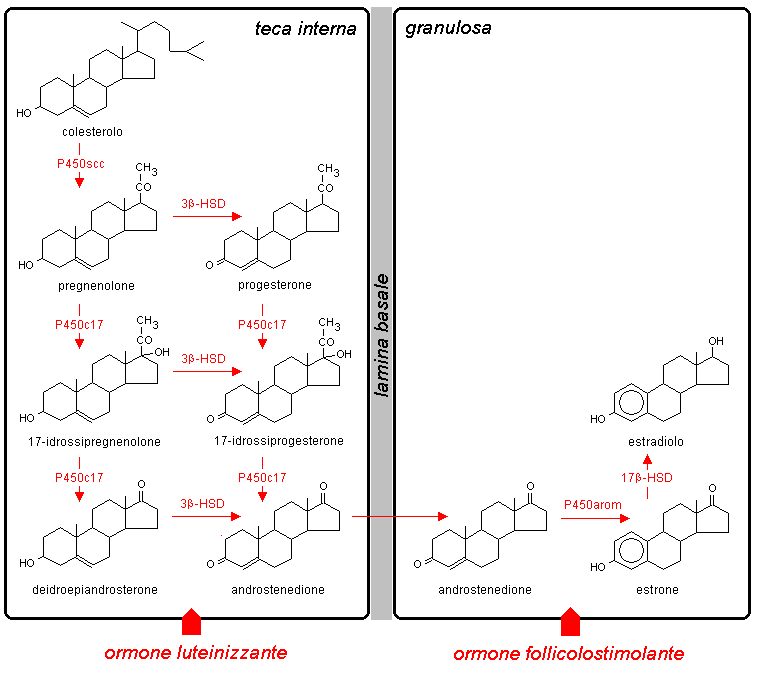
Fig. 10.10. Sistema a 2 cellule / 2 gonadotropine, utilizzato per la sintesi dell’estradiolo a livello dell’ovaio. Il follicolo ooforo comprende due compartimenti cellulari distinti, costituiti rispettivamente dalle cellule della teca e dalle cellule della granulosa, che formano tuttavia un unico sistema biochimico integrato. Il principale steroide prodotto dalle cellule della granulosa è l’estradiolo. La sintesi di questo ormone richiede la produzione di androstenedione da parte delle adiacenti cellule della teca e il suo trasporto nelle cellule della granulosa attraverso la lamina basale. Il processo è sotto il controllo dell’ormone luteinizzante (LH), che agisce sulle cellule della teca, e dell’ormone follicolostimolante, che agisce sulla granulosa. P450scc: enzima di scissione della catena laterale del colesterolo; P450c17: 17α-idrossilasi / 17,20 liasi; P450arom: aromatasi; 3β-HSD: 3β-idrossisteroide deidrogenasi / Δ5-4 isomerasi; 17β-HSD: 17β-idrossisteroide deidrogenasi.
La sintesi degli steroidi nell’ovario è controllata da due gonadotropine, l’ormone luteinizzante (LH) e l’ormone follicolo-stimolante (FSH). Entrambi questi ormoni glicoproteici ipofisari sono eterodimeri costituiti da una subunità α di 92 aminoacidi, pressoché identica a quella presente nell’ormone tireotropo (vedi Par 10.4) e nella gonadotropina corionica (vedi Par. 10.7.1), e da una subunità β di 121 aminoacidi (nel caso del LH) o 111 aminoacidi (nel caso del FSH) che conferisce specificità agli ormoni. L’ormone luteinizzante stimola la produzione di androgeni nelle cellule tecali. L’ormone follicolo-stimolante stimola la crescita del folllicolo e la proliferazione delle cellule della granulosa ed incrementa l’attività dell’aromatasi.
Le cellule deputate alla sintesi delle gonadotropine costituiscono il 7-15%
delle cellule dell’adenoipofisi. Circa il 70% di queste cellule sintetizza sia
LH che FSH, mentre le rimanenti sono specifiche per solo uno dei due ormoni.
L’increzione
di LH e FSH è indotta dall’ormone
rilasciante le gonadotropine (GnRH), un decapeptide prodotto a livello
ipotalamico e riversato nel circolo portale ipofisario
![]() . L’ormone luteinizzante viene accumulato all’interno
dei granuli delle cellule dell’adenoipofisi
e liberato sotto stimolazione del GnRH; l’ormone follicolo-stimolante non si
accumula nei granuli e viene perciò in gran parte liberato immediatamente dopo la sintesi,
che è regolata dal GnRH. Sia nell’ipofisi che nel siero sono
presenti diverse isoforme di LH e FSH che differiscono per il contenuto in
carboidrati. Le isoforme più basiche di LH e FSH hanno una maggiore efficacia
biologica, quando vengono saggiate in vitro, ma una vita media più breve in
circolo.
. L’ormone luteinizzante viene accumulato all’interno
dei granuli delle cellule dell’adenoipofisi
e liberato sotto stimolazione del GnRH; l’ormone follicolo-stimolante non si
accumula nei granuli e viene perciò in gran parte liberato immediatamente dopo la sintesi,
che è regolata dal GnRH. Sia nell’ipofisi che nel siero sono
presenti diverse isoforme di LH e FSH che differiscono per il contenuto in
carboidrati. Le isoforme più basiche di LH e FSH hanno una maggiore efficacia
biologica, quando vengono saggiate in vitro, ma una vita media più breve in
circolo.
Il GnRH è liberato in modo discontinuo ad intervalli di circa un impulso ogni
1-8 ore
![]() . La
frequenza e l’ampiezza degli impulsi è cruciale per l’increzione ipofisaria
delle gonadotropine. L’increzione di LH è generalmente sincrona a quella del GnRH.
Impulsi a più alta frequenza favoriscono l’increzione di LH, mentre impulsi a
più bassa frequenza favoriscono l’increzione di FSH. Una stimolazione
continua o con impulsi di GnRH ad un frequenza più elevata di quella fisiologica
desensibilizza l’ipofisi e sopprime la produzione di LH.
. La
frequenza e l’ampiezza degli impulsi è cruciale per l’increzione ipofisaria
delle gonadotropine. L’increzione di LH è generalmente sincrona a quella del GnRH.
Impulsi a più alta frequenza favoriscono l’increzione di LH, mentre impulsi a
più bassa frequenza favoriscono l’increzione di FSH. Una stimolazione
continua o con impulsi di GnRH ad un frequenza più elevata di quella fisiologica
desensibilizza l’ipofisi e sopprime la produzione di LH.
Basse dosi di estrogeni inibiscono l’increzione di LH e FSH in quanto sia
diminuiscono l’ampiezza degli impulsi di GnRH da parte dell’ipotalamo, sia
deprimono direttamente l’ipofisi attraverso un meccanismo di
feedback negativo. La somministrazione di progesterone rallenta invece
principalmente la
frequenza degli impulsi di GnRH. Tuttavia, oltre ad esercitare un’azione
inibente, in opportune condizioni gli estrogeni esercitano anche un feedback positivo
sull’asse ipotalamico-ipofisario, che è essenziale
per indurre l’aumento preovulatorio di increzione dell’ormone luteinizzante. Infatti,
un progressivo aumento dei livelli di estradiolo, associato ad una
rapida frequenza di stimolazione con GnRH, determina una sensibilizzazione dell’ipofisi al GnRH
attraverso un aumento dei recettori specifici. La modulazione della risposta
ipofisaria al GnRH può risentire inoltre dell’azione di numerosi altri fattori (come ad
esempio le inibine, le attivine e la follistatina
![]() ).
).
10.6.1. Il ciclo mestruale
La durata del ciclo mestruale è normalmente di 28 + 7 giorni. Se la durata del ciclo è inferiore a 21 giorni si parla di polimenorrea, mentre se è superiore a 35 giorni si parla di oligomenorrea (o di amenorrea se supera 180 giorni). Per la maggior parte della vita riproduttiva, vi sono poche variazioni nella durata del ciclo. Queste variazioni sono più frequenti nel periodo immediatamente susseguente al menarca o in prossimità della menopausa, principalmente a causa di fluttuazioni nella durata della fase follicolare. Il ciclo mestruale viene usualmente diviso in quattro fasi e il suo inizio viene fatto coincidere con l’inizio delle mestruazioni (Fig. 10.11).
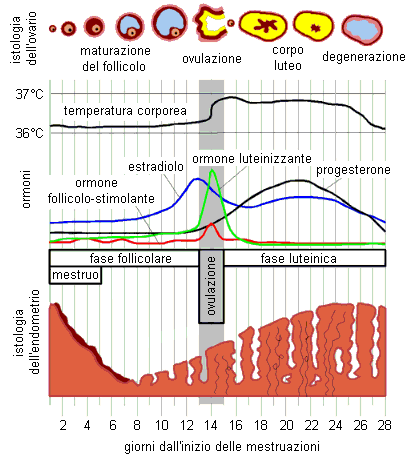
Fig. 10.11. Variazioni cicliche dei livelli
ormonali e della morfologia dell’ovaio e dell’endometrio durante il ciclo
mestruale
![]() .
I valori riportati sono quelli mediamente osservabili nella popolazione
femminile in età fertile. La durata del ciclo e
l’ampiezza delle variazioni indicate in figura variano a seconda del soggetto
esaminato.
.
I valori riportati sono quelli mediamente osservabili nella popolazione
femminile in età fertile. La durata del ciclo e
l’ampiezza delle variazioni indicate in figura variano a seconda del soggetto
esaminato.
10.6.1a. Flusso mestruale e transizione luteo-follicolare
La durata delle mestruazioni è generalmente di 3-5 giorni; è superiore a 7 giorni in caso metrorragia. La quantità del flusso mestruale è 50 + 30 mL. Normalmente il flusso è inizialmente moderato e aumenta progressivamente per poi diminuire. Un sanguinamento improvviso e/o un flusso superiore a 80 mL si hanno in caso di menorragia o ipermenorrea, mentre quantità inferiori a 20 mL si hanno in caso di ipomenorrea. Il sangue è reso incoagulabile per la presenza di plasmina (vedi Par. 7.3.2) ed è frequentemente misto a frustoli di tessuto di derivazione endometriale.
La fase di transizione luteo-follicolare è caratterizzata da un aumento di FSH che inizia generalmente già prima dell’inizio delle mestruazioni ed è essenziale per il reclutamento e lo sviluppo di una nuova coorte di follicoli. L’aumento di FSH è reso possibile dalla diminuzione del livello di estrogeni e si correla ad un aumento di frequenza degli impulsi di LH e di GnRH che passano da un impulso ogni 4-8 ore a un impulso ogni 90-100 minuti.
10.6.1b. Fase follicolare
La prima parte della fase follicolare è caratterizzata da una marcata diminuzione della frequenza degli impulsi di GnRH durante il sonno, probabilmente per assicurare una adeguata produzione di FSH. Nella seconda parte della fase follicolare la frequenza degli impulsi di GnRH aumenta ulteriormente fino a raggiungere un impulso all’ora. L’ampiezza degli impulsi incretori di LH diminuisce marcatamente in questo periodo sia perché è aumentata la loro frequenza, sia in risposta ad una azione inibitoria esercitata dall’estradiolo prodotto dai follicoli in via di sviluppo.
L’aumento di FSH determina l’iniziale sviluppo di 5-7 follicoli che entrano in competizione fra loro. A seguito dell’increzione di LH, i follicoli iniziano a produrre estradiolo. Questo ormone, assieme a fattori inibenti prodotti dal follicolo dominante, inibisce a sua volta la produzione di FSH e determina l’atresia di tutti i follicoli ad eccezione di uno che continua il suo sviluppo ed è destinato all’ovulazione.
10.6.1c. Fase ovulatoria
In risposta all’aumento esponenziale dei livelli di estradiolo nella parte finale della fase follicolare, l’increzione di LH aumenta di circa dieci volte in un periodo di 2-3 giorni, mentre l’increzione di FSH aumenta di circa quattro volte. Durante la fase ovulatoria non si osserva alcun aumento della frequenza o dell’ampiezza degli impulsi di GnRH. Alla fine della fase ovulatoria, l’ampiezza e la frequenza degli impulsi di GnRH e di LH diminuisce nuovamente fino a raggiungere circa un impulso ogni 70 minuti, a causa dell’aumento del livello di progesterone.
L’aumento di increzione di LH a metà ciclo è essenziale perché avvenga la maturazione finale del follicolo e la sua rottura, che normalmente si realizza 36 ore dopo. L’ovulazione è accompagnata a cambiamenti caratteristici del muco cervicale e, a volte, a un dolore caratteristico che può durare diverse ore (Mittelschmerz).
10.6.1d. Fase luteinica
Dopo l’ovulazione, le pareti del follicolo vanno incontro ad un edema sierofibroso, talvolta accompagnato ad una scarsa perdita ematica e alla formazione di un coagulo. Nelle 24 ore successive, la granulosa parietale è invasa da capillari sanguigni; dalla teca interna migrano nella granulosa grosse cellule stellari con citoplasma intensamente eosinofilo (cellule K di Hertig), mentre le cellule della granulosa entrano in attiva proliferazione e si trasformano in cellule luteiniche. Questa nuova formazione, del diametro di 17-20 mm, prende il nome di corpo luteo ed ha il carattere di ghiandola endocrina transitoria capace di elaborare progesterone ed estrogeni. Durante questa fase, la temperatura corporea della donna aumenta di 0,25-0,50° C.
Il corpo luteo gioca un ruolo fondamentale nel preparare l’utero all’eventuale annidamento dell’uovo fecondato (fase secretiva del ciclo funzionale dell’endometrio) e nel sostenere la gravidanza. Se è avvenuta la fecondazione, il corpo luteo si ipertrofizza ancora di più in risposta all’increzione di gonadotropina corionica da parte delle cellule trofoblastiche. Se l’ovocito non viene fecondato, insorgono invece rapidi fenomeni di regressione con degenerazione grassa delle cellule luteiniche e cessazione dell’attività funzionale endocrina che porta alla disintegrazione e allo sfaldamento dello strato funzionale dell’endometrio.
Durante la fase luteinica, l’increzione di FSH è inibita dall’estradiolo, mentre la frequenza degli impulsi di GnRH e di LH continua a rallentare fino a raggiungere, in conseguenza dell’aumento del livello di progesterone, una frequenza pari ad un impulso ogni 4-8 ore.
10.6.2. Metodi di determinazione
Il dosaggio delle gonadotropine, degli estrogeni e dei progestinici si basa
generalmente sull’utilizzo di metodiche RIA
![]() .
Queste metodiche non sono tuttavia sufficientemente sensibili quando è
necessario dosare bassi livelli di gonadotropine; in questi casi è preferibile
perciò ricorrere a metodiche che prevedono l’utilizzo di un secondo anticorpo
legato ad una fase solida per separare i reagenti in eccesso (metodiche ELISA o IRMA
.
Queste metodiche non sono tuttavia sufficientemente sensibili quando è
necessario dosare bassi livelli di gonadotropine; in questi casi è preferibile
perciò ricorrere a metodiche che prevedono l’utilizzo di un secondo anticorpo
legato ad una fase solida per separare i reagenti in eccesso (metodiche ELISA o IRMA
![]() ). Per dosare
basse concentrazioni di estrogeni, è necessario estrarre gli analiti in solvente
organico e separare gli eventuali steroidi interferenti mediante cromatografia.
). Per dosare
basse concentrazioni di estrogeni, è necessario estrarre gli analiti in solvente
organico e separare gli eventuali steroidi interferenti mediante cromatografia.
10.6.3. Preparazione del campione ed intervalli di riferimento
La concentrazione delle gonadotropine nel siero è generalmente valutata in rapporto a preparazioni standard di riferimento ed espressa in unità internazionali, tuttavia la disponibilità di nuovi standard di gonadotropine ricombinanti può permettere di quantizzare queste proteine anche in unità di massa.
Nella donna in età fertile, la concentrazione di LH e FSH, misurata con
metodi RIA, è compresa fra 4 e 15 mU/mL; il rapporto LH/FSH è circa 1 durante la
fase follicolare, mentre aumenta durante l’ovulazione. Prima della pubertà, i
livelli delle gonadotropine sono più bassi e il rapporto LH/FSH è inferiore a 1.
In prossimità della menopausa, il livello di FSH aumenta, mentre quello di LH
rimane nei limiti normali. Dopo la menopausa, entrambe le gonadotropine
aumentano di concentrazione, ma in età più avanzata i valori di LH diminuiscono
nuovamente fino a raggiungere livelli prossimi a quelli di una donna in età
fertile. Alti livelli di LH e un alto rapporto LH/FSH (> 2) si riscontrano in
donne con sindrome da ovario policistico
![]() . Alti livelli di LH e FSH si hanno in
pazienti con disgenesia gonadica, mentre bassi livelli di LH e FSH possono
essere associati a disordini ipotalamo-ipofisari. I tumori ipofisari raramente
portano ad un aumento di gonadotropine.
. Alti livelli di LH e FSH si hanno in
pazienti con disgenesia gonadica, mentre bassi livelli di LH e FSH possono
essere associati a disordini ipotalamo-ipofisari. I tumori ipofisari raramente
portano ad un aumento di gonadotropine.
Prove funzionali per la valutazione dell’attività gonadotropinica possono essere eseguite somministrando GnRH sintetico o citrato di clomifene. Il test che utilizza GnRH sintetico (25-100 mg endovena) serve per differenziare un deficit ipotalamico da un deficit ipofisario e per svelare ritardi secretivi in età pubere. Il test che utilizza il clomifene (100 mg per os al giorno per 5-9 giorni) sfrutta la proprietà di questo estrogeno non steroideo di interferire con i recettori estrogenici presenti a livello ipotalamico/ipofisario e di determinare, in tal modo, un incremento dei livelli di FSH ed LH.
La concentrazione di estradiolo nel siero è di 20-80 pg/mL durante la parte iniziale della fase follicolare del ciclo mestruale con un picco di 200-500 pg/mL nel periodo che precede l’ovulazione. Durante la fase luteinica, l’estradiolo sierico è pari a 60-200 pg/mL. Nelle ragazze prepuberi, i livelli di estradiolo sono inferiori a 20 pg/mL; simili valori sono riscontrabili in donne che hanno superato la menopausa. Livelli relativamente elevati di estrone si osservano in donne sotto terapia orale con estrogeni.
Nel siero dei maschi, l’estradiolo è inferiore a 40 pg/mL, mentre l’estrone è inferiore a 60 pg/mL. A differenza del testosterone (vedi Par. 10.8.2), il livello di estradiolo sierico si mantiene costante con l’età; un moderato aumento di estrogeni è riscontrabile nei soggeti obesi, mentre aumenti più consistenti si possono osservare in diverse malattie riguardanti i testicoli o altri organi.
Il progesterone sierico è inferiore a 1,5 ng/mL durante la fase follicolare del ciclo mestruale, inizia ad aumentare nel periodo che precede l’ovulazione e raggiunge un massimo (7-15 ng/mL) 6-8 giorni dopo l’ovulazione. In caso di gravidanza, il livello di progesterone sale progressivamente, raggiungendo 40 ng/mL alla fine del primo trimestre e 150 ng/mL a termine.
La determinazione del livello di progesterone nel siero è utile per lo studio del ciclo mestruale e per verificare l’efficacia di terapie volte ad indurre l’ovulazione. Bassi livelli di progesterone (< 10 ng/mL) tra la sesta e l’ottava settimana di gestazione sono indice di una gravidanza intrauterina anormale o di una gravidanza ectopica.
10.7. ORMONI DELLA PLACENTA
La placenta non solo provvede allo scambio di alimenti, gas e prodotti di escrezione fra madre e feto, ma si comporta anche come un importante organo endocrino. La placenta produce un gran numero di proteine (inclusi i fattori di crescita e le citochine), neuropeptidi, monoamine e steroidi. Molti degli ormoni placentari sono simili a quelli prodotti dalla donna non gravida. Essi sono riversati in grande quantità nel sangue materno e si legano ai recettori ormonali della madre modificandone i meccanismi omeostatici per soddisfare le esigenze metaboliche e nutrizionali del feto in rapida crescita.
10.7.1. Ormoni di tipo pituitarico
La gonadotropina corionica umana (hCG) è una glicoproteina di 40 kDa
biologicamente ed immunologicamente simile all’ormone luteinizzante ipofisario
(LH). E’ un eterodimero costituito da una subunità α,
pressoché identica a quella presente negli ormoni glicoproteici ipofisari
(ormone follicolo-stimolante, FSH; ormone luteinizzante, LH; ormone tireotropo,
TSH), e da una subunità β, che conferisce
all’ormone la sua specificità (vedi Par 10.1). La
produzione della godanotropina corionica da parte delle cellule
trofoblastiche della blastocisti inizia immediatamente prima dell’impianto e
continua per tutto il proseguo della gravidanza. La placenta non è la sola
sorgente dell’ormone, che viene sintetizzato anche dai reni e dall’ipofisi del
feto. L’ormone hCG prolunga la vita del corpo luteo, che viene trasformato in
corpo luteo gravidico, e stimola la produzione di progesterone, regolando la
steroidogenesi placentare (vedi Par. 10.7.2). Altri bersagli dell’ormone sono la ghiandola tiroidea
della madre e i surreni e i testicoli del feto. La produzione di godanotropina
corionica è, a sua volta, controllata dai livelli di progesterone, attivina, inibina
![]() ed ormone rilasciante le gonadotropine (GnRH, vedi Par
10.1), che sono sintetizzati dalla placenta stessa. In una gravidanza
normale, la gonadotropina corionica è evidenziabile nel sangue e nell’urina della
madre dopo circa 8 giorni dalla ovulazione e dopo un solo giorno dall’impianto. Il
suo livello ematico raddoppia ogni due-tre giorni, raggiunge un massimo dopo
60-90 giorni, poi diminuisce fino a raggiungere un plateau che viene
mantenuto per il resto della gravidanza. La vita media dell’ormone hCG è di
32-37 ore, considerevolmente più alta rispetto a quella del FSH, dell’LH e degli
ormoni steroidei.
ed ormone rilasciante le gonadotropine (GnRH, vedi Par
10.1), che sono sintetizzati dalla placenta stessa. In una gravidanza
normale, la gonadotropina corionica è evidenziabile nel sangue e nell’urina della
madre dopo circa 8 giorni dalla ovulazione e dopo un solo giorno dall’impianto. Il
suo livello ematico raddoppia ogni due-tre giorni, raggiunge un massimo dopo
60-90 giorni, poi diminuisce fino a raggiungere un plateau che viene
mantenuto per il resto della gravidanza. La vita media dell’ormone hCG è di
32-37 ore, considerevolmente più alta rispetto a quella del FSH, dell’LH e degli
ormoni steroidei.
La somatomammotropina (hCS), che è denominata anche ormone lattogeno
placentare (hPL), è una proteina costituita da una unica catena di 191 aminoacidi che ha il 96% di omologia con l’ormone somatotropo. Durante la gravidanza,
questo ormone è formato, fino alla sesta settima, dal citotrofoblasto e, in
seguito, dal sinciziotrofoblasto. La quantità di ormone prodotto aumenta con il
crescere delle dimensioni della placenta, dalla quale viene rilasciato
principalmente nel comparto materno (compare nel sangue materno dopo 20-40
giorni di gestazione e raggiunge a termine la concentrazione di 5-10
mg/mL). La concentrazione ematica di hCS aumenta in
condizioni di stress metabolico (digiuno prolungato, ipoglicemia indotta
da insulina), ma un suo deficit isolato, anche se completo, non compromette il decorso
normale della gravidanza
![]() .
.
Le cellule del sinciziotrofoblasto producono anche due ormoni placentari della crescita (hPGH), uno di 26 kDa ed uno di 22 kDa; quest’ultimo differisce dalla somatotropina ipofisaria solo per 13 aminoacidi (vedi Par. 10.2). L’hPGH non entra nel compartimento fetale, mentre è dosabile dopo 20-25 settimane di gravidanza nel sangue materno, dove raggiunge a termine la concentrazione di circa 20 ng/mL, subentrando nelle sue funzioni all’omologo ormone materno. Infatti, durante il primo trimestre di gravidanza, la somatotropina ipofisaria continua ad essere prodotta dalla madre con un andamento pulsatile, ma la sua concentrazione diminuisce nel secondo trimestre e, nel terzo trimestre, l’ormone materno è completamente sostituito da quello fetale che, a differenza del precedente, viene increto dalla placenta in modo continuo. La carenza di hPGH (che sempre si accompagna anche ad una carenza di hCG) determina un grave ritardo nella crescita fetale.
La pro-opiomelanocortina (vedi Par. 10.5.1a) viene
prodotta anch’essa dalla placenta in modo continuo, ma, a differenza di quanto avviene a
livello dell’ipofisi, la pro-opiomelanocortina placentare viene scissa solo in
parte nelle sue componenti
![]() . La pro-opiomelanocortina è
dosabile nel sangue materno dopo il terzo mese, raggiunge un plateau di
circa 300 U/mL a metà gestazione e scompare nuovamente dal circolo subito dopo
il parto. Il livello di pro-opiomelanocortina nel sangue materno non dipende dal livello di
ACTH o di cortisolo, mentre si correla bene con il livello plasmatico
dell’ormone rilasciante la corticotropina (CRH). Il ruolo fisiologico dei
prodotti di idrolisi della pro-opiomelanocortina placentare (corticotropina,
lipotropina, b-endorfina, ormone stimolante i
melanociti) è ancora poco noto.
. La pro-opiomelanocortina è
dosabile nel sangue materno dopo il terzo mese, raggiunge un plateau di
circa 300 U/mL a metà gestazione e scompare nuovamente dal circolo subito dopo
il parto. Il livello di pro-opiomelanocortina nel sangue materno non dipende dal livello di
ACTH o di cortisolo, mentre si correla bene con il livello plasmatico
dell’ormone rilasciante la corticotropina (CRH). Il ruolo fisiologico dei
prodotti di idrolisi della pro-opiomelanocortina placentare (corticotropina,
lipotropina, b-endorfina, ormone stimolante i
melanociti) è ancora poco noto.
La produzione placentare dell’ormone rilasciante la corticotropina (CRH)
inizia dalla settima settimana di gestazione, aumenta progressivamente nei mesi
successivi ed in modo esponenziale nelle ultime 5-7 settimane di gravidanza
![]() . Dopo 15 settimane di gestazione,
il CRH è dosabile nel sangue materno, dove raggiunge a termine la concentrazione
di 1-10 ng/mL. Malgrado la forte increzione di CRH, l’ipofisi materna non è
stimolata a produrre maggiori quantità di ACTH durante la gravidanza in quanto,
da un lato, l’ormone placentare rilasciante la corticotropina è inibito da una
specifica proteina di trasporto presente in largo eccesso nel sangue materno
(almeno nella prima parte della gravidanza), dall’altro lato, la risposta stessa
dell’ipofisi materna agli stimoli risulta attenuata. Al contrario, l’ormone
placentare, che penetra (sebbene in minore quantità) nel torrente circolatorio
fetale, determina un aumento di produzione di ACTH da parte dell’ipofisi fetale
e, di conseguenza, un aumento di cortisolo da parte dei surreni fetali. A sua
volta, il cortisolo stimola la produzione placentare di CRH (l’increzione ipotalamica di CRH nell’adulto è invece inibita dai glicocorticoidi, vedi Par. 10.5.1a)
con un meccanismo di feed-back positivo, che è causa del forte incremento
di CRH nell’ultima parte della gravidanza. Bisogna infine ricordare che il CRH può esercitare una azione diretta sulla steroidogenesi fetale (è 70% meno
potente dell’ACTH nello stimolare la produzione di cortisolo, ma ha la stessa
capacità dell’ACTH nello stimolare la produzione di deidroepiandrosterone
solfato) che è in stretta relazione con la biosintesi degli estrogeni da parte
della placenta, che in tal modo viene stimolata (vedi
Par. 10.7.2).
. Dopo 15 settimane di gestazione,
il CRH è dosabile nel sangue materno, dove raggiunge a termine la concentrazione
di 1-10 ng/mL. Malgrado la forte increzione di CRH, l’ipofisi materna non è
stimolata a produrre maggiori quantità di ACTH durante la gravidanza in quanto,
da un lato, l’ormone placentare rilasciante la corticotropina è inibito da una
specifica proteina di trasporto presente in largo eccesso nel sangue materno
(almeno nella prima parte della gravidanza), dall’altro lato, la risposta stessa
dell’ipofisi materna agli stimoli risulta attenuata. Al contrario, l’ormone
placentare, che penetra (sebbene in minore quantità) nel torrente circolatorio
fetale, determina un aumento di produzione di ACTH da parte dell’ipofisi fetale
e, di conseguenza, un aumento di cortisolo da parte dei surreni fetali. A sua
volta, il cortisolo stimola la produzione placentare di CRH (l’increzione ipotalamica di CRH nell’adulto è invece inibita dai glicocorticoidi, vedi Par. 10.5.1a)
con un meccanismo di feed-back positivo, che è causa del forte incremento
di CRH nell’ultima parte della gravidanza. Bisogna infine ricordare che il CRH può esercitare una azione diretta sulla steroidogenesi fetale (è 70% meno
potente dell’ACTH nello stimolare la produzione di cortisolo, ma ha la stessa
capacità dell’ACTH nello stimolare la produzione di deidroepiandrosterone
solfato) che è in stretta relazione con la biosintesi degli estrogeni da parte
della placenta, che in tal modo viene stimolata (vedi
Par. 10.7.2).
10.7.2. Ormoni steroidei
La placenta è priva dell’enzima bifunzionale 17α-idrossilasi / 17,20-liasi (vedi Par.10.5) e, perciò, non può completare la sintesi degli ormoni steroidei dopo aver trasformato in progesterone il colesterolo fornito dal sangue materno. Per formare gli estrogeni, attraverso l’azione dell’enzima aromatasi di cui è ricca, la placenta utilizza gli androgeni prodotti dal feto e dalla madre, in primo luogo il deidroepiandrosterone solfato prodotto dai surreni fetali. Il deidroepiandrosterone solfato è convertito prima in deidroepiandrosterone libero dalla solfatasi placentare, poi in androstenedione, in testosterone ed infine in estradiolo ed estrone.
Tuttavia, il principale estrogeno che si forma nel corso della gravidanza non
è l’estrone o l’estradiolo, bensì l’estriolo (più del 90% degli estrogeni
urinari in gravidanza è costituito infatti da estriolo). Esso si forma a partire
dal deidroepiandrosterone solfato, prodotto dai surreni fetali, attraverso una
via metabolica utilizzata esclusivamente in gravidanza (Fig.
10.12). Il deidroepiandrosterone solfato è trasformato in 16α-idrossideidroepiandrosterone
solfato nel fegato fetale e, in minor misura, nei surreni fetali. Il 16α-idrossideidroepiandrosterone
solfato raggiunge quindi la placenta, dove viene convertito in 16α-idrossideidroepiandrosterone
libero dalla solfatasi ed infine in estriolo dalla aromatasi. L’estriolo viene
immesso nella circolazione materna e raggiunge il fegato della madre, dove viene
convertito in estriolo solfato, estriolo glucuronato ed estriolo
sulfoglucuronato per l’azione combinata della solfotransferasi
![]() e
della UDP-glucurosiltransferasi.
e
della UDP-glucurosiltransferasi.
Un altro estrogeno placentare, che deriva da precursori fetali, è l’estetrolo che si forma a seguito di una ossidrilazione in posizione 15 del 16α-idrossideidroepiandrosterone solfato.
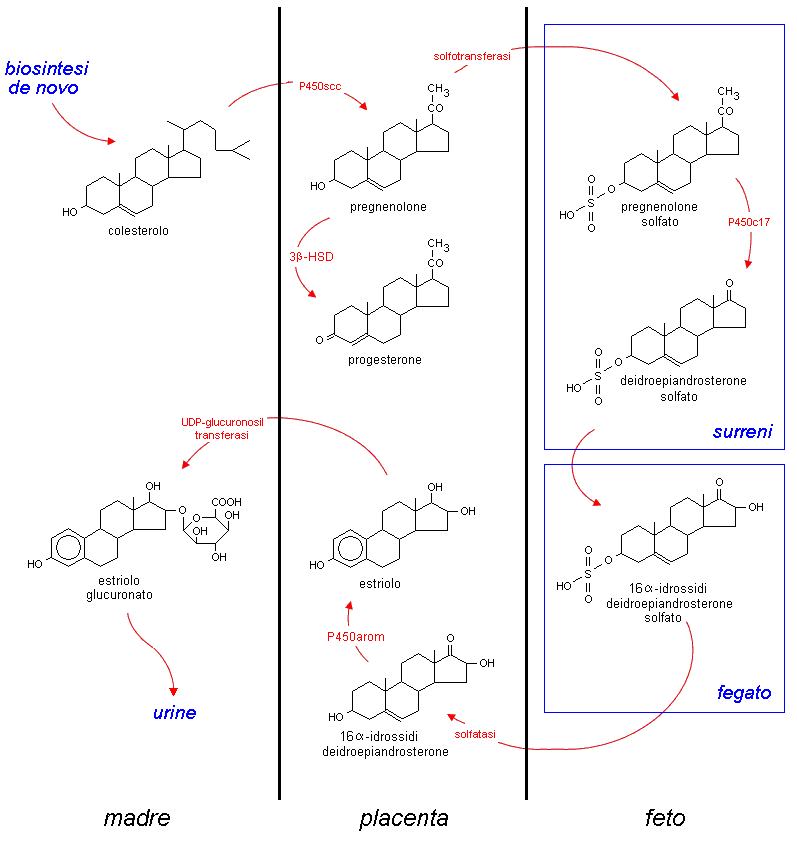
Fig. 10.12. Biosintesi dell’estriolo durante la gravidanza. La placenta è priva dell’enzima 17α-idrossilasi / 17,20-liasi. Per completare la sintesi steroidea, la placenta si avvale del metabolismo della madre e del feto, costituendo una unità metabolica integrata materno-feto-placentare. P450scc: enzima di scissione della catena laterale del colesterolo; P450c17: 17α-idrossilasi / 17,20 liasi; P450arom: aromatasi; 3β-HSD: 3β-idrossisteroide deidrogenasi / Δ5-4 isomerasi; solfatasi: solfatasi steroidea; solfotransferasi: solfotransferasi degli idrossisteroidi.
10.7.3. Metodi di determinazione
Per il dosaggio degli ormoni steroidei e di tipo pituitarico fetali sono generalmente utilizzate metodiche cromatografiche o basate sull’uso di anticorpi specifici (vedi Par. 10.6.2). Sono reperibili in commercio semplici e rapidi test per la diagnosi di gravidanza sotto forma di bastoncini o cartine che possono essere adoperati a domicilio.
10.7.4. Preparazione del campione ed intervalli di riferimento
Metodi che evidenziano la presenza di gonadotropina corionica nel sangue o nelle urine sono comunemente adoperati come test di gravidanza. I comuni test urinari sono qualitativi o semiquantitativi ed hanno una sensibilità pari a 20-100 mU/mL. E’ consigliabile eseguire il saggio sulle prime urine del mattino, in quanto in esse l’ormone è più concentrato; le urine vanno diluite se il loro peso specifico relativo è superiore a 1,015. I test che utilizzano il sangue venoso hanno una maggiore sensibilità (pari a circa 5 mU/mL) e permettono una determinazione quantitativa dell’ormone. Elevati livelli di gonadotropina corionica sono evidenziabili anche in caso di mola idatiforme e di alcuni tumori (corioncarcinoma, teratomi, tumori neuroendocrini).
Il dosaggio simultaneo della concentrazione della gonadotropina corionica,
dell’estriolo non coniugato e dell’α-fetoproteina
![]() nel
plasma materno costituisce il Tri-test (test triplo di
Kettering o di Bart), che è utilizzato nel secondo trimestre di gravidanza
come test di screening prenatale per la valutazione del rischio di sindrome di
Down (vedi Par. 14.6.2). Poiché
i livelli plasmatici degli analiti si distribuiscono, nei controlli e nelle
pazienti con sindrome di Down, su ampi intervalli di concentrazione in gran
parte sovrapposti (vedi Fig. 10.13), il Tri-test
consente di ottenere un valore di predittività non superiore al 75% con una
percentuale di falsi positivi pari a circa 8%. La predittività del test
può essere incrementata dosando la subunità β libera
della gonadotropina corionica o associando il Tri-test al dosaggio
della inibina A
nel
plasma materno costituisce il Tri-test (test triplo di
Kettering o di Bart), che è utilizzato nel secondo trimestre di gravidanza
come test di screening prenatale per la valutazione del rischio di sindrome di
Down (vedi Par. 14.6.2). Poiché
i livelli plasmatici degli analiti si distribuiscono, nei controlli e nelle
pazienti con sindrome di Down, su ampi intervalli di concentrazione in gran
parte sovrapposti (vedi Fig. 10.13), il Tri-test
consente di ottenere un valore di predittività non superiore al 75% con una
percentuale di falsi positivi pari a circa 8%. La predittività del test
può essere incrementata dosando la subunità β libera
della gonadotropina corionica o associando il Tri-test al dosaggio
della inibina A
![]() (test
quadruplo).
(test
quadruplo).
Fig. 10.13. Distribuzione di frequenza dei livelli di gonadotropina corionica (hCG), estriolo non coniugato (uE3) e α-fetoproteina nel plasma di donne al secondo trimestre di gravidanza. Le curve si riferiscono a controlli sani (croci blu) e a pazienti con feto affetto da sindrome di Down (pallini rossi).
10.8. ORMONI DELLE GONADI MASCHILI
La sintesi del testosterone avviene nelle cellule di Leydig, che sono situate
nel compartimento interstiziale che costituisce circa il 5% del volume del
testicolo maturo (Fig. 10.14). Il colosterolo necessario per
la sintesi è in gran parte formato de novo a partire dall’acetato, ma può anche
provenire dalle riserve cellulari o dalle LDL presenti in circolo (vedi
Par. 3). La conversione del colesterolo in
testosterone richiede l’intervento, nei mitocondri, dell’enzima di scissione
della catena laterale del colesterolo e, nei microsomi, dell’enzima 17α-idrossilasi
/ 17-20 liasi (vedi Par. 10.5). Gli altri enzimi microsomiali,
che intervengono nelle sintesi, sono la 3β-idrossisteroide
deidrogenasi / Δ5-4 isomerasi
![]() e la
17β-idrossisteroide
deidrogenasi
e la
17β-idrossisteroide
deidrogenasi
![]() . Il
trasferimento del colesterolo nei mitocondri è indotto dal cAMP ed è controllato da alcune proteine chiave, la
StAR (Steroidogenic Acute Regulatory Protein) e la
proteina trasportatriche degli steroli.
. Il
trasferimento del colesterolo nei mitocondri è indotto dal cAMP ed è controllato da alcune proteine chiave, la
StAR (Steroidogenic Acute Regulatory Protein) e la
proteina trasportatriche degli steroli.
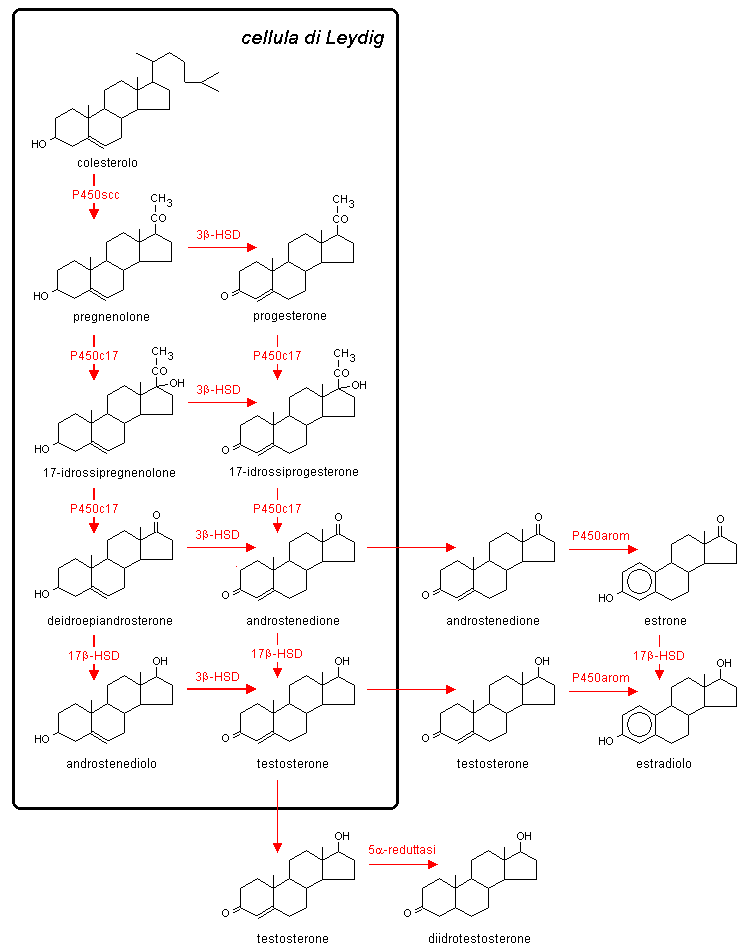
Fig. 10.14. Biosintesi degli ormoni sessuali maschili. La figura mostra le vie di sintesi del testosterone nelle cellule di Leydig. La sintesi del testosterone attraverso la via che passa per il pregnenolone, il deidroepiandrosterone e l’androstenediolo (in grossetto) è, nella specie umana, preferita rispetto alla via che passa per il progesterone e l’androstenedione. Il testosterone e il suo metabolita, diidrotestosterone, possono esercitare i loro effetti biologici direttamente, mediante il legame ai recettori periferici per gli androgeni, o indirettamente dopo aromatizzazione del testosterone a estradiolo, mediante il legame ai recettori periferici per gli estrogeni. P450scc: enzima di scissione della catena laterale del colesterolo; P450c17: 17α-idrossilasi / 17,20 liasi; P450arom: aromatasi; 3β-HSD: 3β-idrossisteroide deidrogenasi / Δ5-4 isomerasi; 17β-HSD: 17β-idrossisteroide deidrogenasi; 5α-reduttasi: 5α-reduttasi.
Il testosterone è increto nel maschio ad alte concentrazioni in tre periodi
successivi: nel primo trimestre di vita intrauterina (fra l’ottava e la
diciottesima settimana) in coincidenza con la differenziazione del tratto
genitale, nel periodo neonatale (fra il secondo e il terzo mese di vita) e, con
continuità, dopo la pubertà per mantenere lo stato di virilizzazione. Dopo la
pubertà, circa il 95% del testosterone in circolo è prodotto dai testicoli,
mentre il rimanente deriva dall’androstenedione e dall’epiandrosterone,
prodotti dal surrene (vedi Par. 10.5
![]() ).
).
L’increzione del testosterone è regolata dall’ormone ipofisario luteinizzante
(LH, vedi Par. 10.6) che stimola la steroidogenesi delle cellule di Leydig
aumentando la disponibilità di colesterolo, favorendo il suo trasporto nel
mitocondrio ed attivando gli enzimi della via biosintetica. I recettori per l’LH
sono situati sulla superficie delle cellule di Leydig ed utilizzano meccanismi
di trasduzione del segnale che coinvolgono il cAMP e gli ioni calcio in processi
di fosforilazione delle proteine attraverso chinasi specifiche. L’ormone luteinizzante è tipicamente prodotto dall’ipofisi in modo discontinuo e la sua
concentrazione ematica presenta picchi che si ripetono nel tempo circa uno
all’ora; ciò permette il normale funzionamento delle cellule di Leydig, che
divengono, invece, insensibili all’LH quando sono esposte in modo continuativo a
questo ormone. L’increzione di testosterone da parte delle cellule di Leydig è
inoltre influenzata indirettamente da fattori che variano il flusso ematico o
che agiscono attraverso le cellule di Sertoli, come l’attivina, l’inibina
![]() ,
l’ormone follicolo-stimolante, la prolattina, le prostaglandine E2 e
F2a, la somatotropina ed i fattori insulino-simili. Il testosterone,
a sua volta, inibisce la produzione dei fattori rilascianti le gonadotropine (GnRH)
riducendo di conseguenza l’increzione di LH da parte dell’adenoipofisi. Questo
meccanismo di retroreazione (feed-back) negativa coinvolge ricettori a
cui si legano il testosterone o l’estradiolo; quest’ultimo composto deriva dalla aromatizzazione
del testosterone stesso.
,
l’ormone follicolo-stimolante, la prolattina, le prostaglandine E2 e
F2a, la somatotropina ed i fattori insulino-simili. Il testosterone,
a sua volta, inibisce la produzione dei fattori rilascianti le gonadotropine (GnRH)
riducendo di conseguenza l’increzione di LH da parte dell’adenoipofisi. Questo
meccanismo di retroreazione (feed-back) negativa coinvolge ricettori a
cui si legano il testosterone o l’estradiolo; quest’ultimo composto deriva dalla aromatizzazione
del testosterone stesso.
In condizioni fisiologiche, il testosterone presente in circolo è legato per il 60-70% ad una proteina dimerica di 95 kDa prodotta dal fegato (SHBG, Sex Hormone-Binding Globulin) e, in minor misura, ad altre proteine a cui è meno affine (albumina, α1-glicoproteina acida, transcortina). Il rimanente 1-2% dell’ormone è presente nel plasma in forma libera, biologicamente attiva. Il destino metabolico del testosterone è di essere trasformato in altri prodotti bioattivi o di essere inattivato ed escreto con la bile o le urine.
Circa il 4% del testosterone circolante è trasformato dalla 5α-reduttasi in un androgeno più potente, il diidrotestosterone. L’isoenzima tipo 1 della 5α-reduttasi è presente in vari tessuti (fegato, reni, pelle, cervello), mentre l’isoenzima tipo 2 è espresso prevalentemente nella prostata. Durante la vita fetale, l’isoenzima tipo 2 ha importanza nel dirigere lo sviluppo del seno urogenitale in senso maschile. I soggetti con deficit congenito dell’isoenzima tipo 2 della 5α-reduttasi hanno perciò genitali ambigui e possono essere scambiati per femmine in età infantile, ma alla pubertà subiscono una forte virilizzazione (inclusa la crescita peninea) e sviluppano occasionalmente un orientamento sessuale di tipo maschile, pur rimanendo la loro prostata rudimentale.
Circa lo 0,2% del testosterone è trasformato in estradiolo dall’enzima aromatasi (nel maschio adulto, circa l’80% dell’estradiolo circolante deriva da questa via). L’estradiolo interagisce con i recettori per gli estrogeni e contribuisce nel soggetto di sesso maschile alla maturazione di vari tessuti, in particolare il tessuto osseo.
Il testosterone è trasformato in metaboliti inattivi dal fegato, reni, intestino, muscoli e tessuto adiposo. L’inattivazione avviene principalmente nel fegato ad opera delle ossidasi della famiglia del citocromo P450 3A e comporta la formazione di derivati ossidati che vengono successivamente coniugati all’acido glucuronico ed escreti prevalentemente con le urine.
10.8.1. Metodi di determinazione
Il dosaggio del testosterone totale e della proteina trasportatrice (SHBG)
utilizza metodi immunologici che sono generalmente basati sulla
competizione, rispetto ad un numero limitato di siti anticorpali, del campione
in esame con una preparazione standard marcata con un isotopo
radioattivo (RIA), con un fluoroforo (FPIA) o con un enzima (EMIT)
![]() .
.
La percentuale di testosterone libero può essere determinata mediante equilibrio di dialisi, lasciando equilibrare una piccola quantità nota di testosterone radioattivo fra i due compartimenti separati dalla membrana semipermeabile. Un altro metodo è quello di calcolare la concentrazione di testosterone libero dalla concentrazioni di testosterone totale, SHBG ed albumina e dalle rispettive costanti di legame. Una misura diretta della concentrazione di testosterone libero può essere ottenuta utilizzando un metodo immunologico basato sulla competizione, rispetto ad un numero limitato di siti anticorpali, del campione in esame con una preparazione standard di analogo del testosterone con bassa affinità per SHBG ed albumina.
Il testosterone biodisponibile è costituito dalla somma del testosterone libero e del testosterone legato all’albumina. La percentuale di testosterone biodisponibile è determinata mediante l’aggiunta di una piccola quantità di testosterone radioattivo al siero e la successiva precipitazione di SHBG in solfato d’ammonio.
L’indice di testosterone libero è dato dal rapporto fra le concentrazioni di testosterone totale e di SHBG, entrambe espresse in nmol/L. L’indice è utilizzato nel caso di campioni di sangue prelevati a donne, mentre ha meno valore nei maschi in quanto in costoro gran parte del testosterone è generalmente in forma legata
10.8.2. Preparazione del campione ed intervalli di riferimento
Plasma e siero sono i campioni di elezione per il dosaggio degli androgeni. Il testosterone è presente anche nella saliva ad una concentrazione paragonabile a quella del testosterone libero nel siero, ma il suo dosaggio richiede un accurato controllo di qualità in quanto può essere soggetto a un maggior numero di artefatti.
L’increzione di testosterone è discontinua con una frequenza grossomodo oraria, ma la concentrazione del testosterone nel sangue periferico non subisce oscillazioni significative a breve termine in quanto gli impulsi incretori sono di breve durata ed ampiezza. Il livello di testosterone nel sangue periferico segue invece una oscillazione giornaliera con un massimo alle prime ore del mattino ed un minimo durante le prime ore del sonno, quando è ad un livello più basso di circa il 15%. Per tale motivo, generalmente si raccomanda di prelevare il sangue per il dosaggio di questo ormone fra le ore 8 e le ore 10.
La concentrazione totale del testosterone nel siero subisce un incremento al secondo-terzo mese di vita, quando raggiunge i valori di 200-300 ng/dL, diminuisce dopo il quarto-sesto mese a livelli bassissimi per risalire nuovamente con l’inizio della pubertà. I valori tipici dell’adulto (300-1000 ng/dL) sono raggiunti verso i 16 anni. La concentrazione di testosterone biodisponibile è 66-417 ng/dL, mentre la concentrazione di testosterone libero è 50-210 pg/dL.
La concentrazione plasmatica di SHBG è di 20-100 nmol/L nei maschi adulti. Negli anziani la concentrazione plasmatica di SHBG è più alta di circa il 75% e, di conseguenza, la concentrazione di testosterone libero è circa un terzo di quella di un soggetto in giovane età.
Prove funzionali possono essere eseguite somministrando gonadotropina corionica umana (hCG), ormone rilasciante le gonadotropine (GnRH) o inibitori specifici. La stimolazione con hCG (1500 UI intramuscolo ogni secondo giorno per 7 giorni) fallisce in caso di un difetto nella biosintesi del testosterone o di una sindrome da insensibilità agli androgeni, mentre può stimolare il rilascio di testosterone in ragazzi con criptorchidia bilaterale; soggetti con deficit di gonadotropine rispondono poco alla somministrazione di hCG per una perdita di sensibilità delle cellule di Leydig. La stimolazione con GnRH (100 pg indovena), introdotta per differenziare gli ipogonadismi dovuti a disfunzione ipofisaria da quelli dovuti a disfunzione ipotalamica, si è rivelata poco utile in quanto nel secondo caso le cellule ipofisarie possono essere desensibilizzate e quindi iporeattive. La somministrazione di inibitori specifici, che agiscono sulla sintesi degli steroidi o sui loro recettori periferici, sblocca il feedback negativo esercitato da queste sostanze sull’ipotalamo e permette di studiare l’integrità funzionale dell’asse ipotalamico-ipofisario.
10.9. ORMONI COINVOLTI NELLA REGOLAZIONE DEL METABOLISMO OSSEO
La regolazione del metabolismo minerale delle ossa è sotto il controllo di quattro principali ormoni (paratormone, proteina correlata al paratormone, calcitonina e 1α,25-diidrossi-vitamina D) che intervengono a livello di tre organi bersaglio (ossa, rene e intestino) nel controllo dell’omeostasi del calcio, fosforo e magnesio. Altri ormoni che intervengono sul metabolismo osseo sono gli androgeni e gli estrogeni, che contribuiscono a mantenere la massa scheletrica, l’insulina, l’ormone della crescita e gli ormoni tiroidei, che promuovono la crescita e la maturazione ossea, ed i glicocorticoidi, che esercitano un’azione negativa su tutte le funzioni scheletriche.
10.9a. Paratormone
Il paratormone (PTH,
parathyroid hormone) è un
peptide di 84 aminoacidi prodotto dalle ghiandole paratiroidi, che sono
presenti generalmente in numero di quattro a ridosso della superficie posteriore
dei lobi tiroidei o in posizione ectopica fra la tiroide e il
mediastino. La trascrizione del gene relativo a questo ormone è inibita
dalla 1α,25-diidrossi-vitamina D (vedi Par.
10.9d) e dagli ioni calcio. Anche l’increzione dell’ormone maturo
![]() , che è contenuto nei granuli
delle cellule
paratiroidee, è inibita da alti livelli plasmatici di calcio ionizzato, mentre è
favorita da bassi livelli di questo catione. Il magnesio esercita un effetto
simile al calcio, ma una prolungata deplezione di magnesio ha un effetto
inibente sulla produzione del paratormone. Il fosfato inorganico regola l’increzione
del paratormone sia direttamente ad alte concentrazioni, sia indirettamente inibendo la sintesi della
1α,25-diidrossi-vitamina D.
, che è contenuto nei granuli
delle cellule
paratiroidee, è inibita da alti livelli plasmatici di calcio ionizzato, mentre è
favorita da bassi livelli di questo catione. Il magnesio esercita un effetto
simile al calcio, ma una prolungata deplezione di magnesio ha un effetto
inibente sulla produzione del paratormone. Il fosfato inorganico regola l’increzione
del paratormone sia direttamente ad alte concentrazioni, sia indirettamente inibendo la sintesi della
1α,25-diidrossi-vitamina D.
Il paratormone ha una emivita inferiore a 5 minuti in quanto viene rapidamente idrolizzato, principalmente a livello del fegato e del rene. I principali frammenti che derivano dall’idrolisi del paratormone sono il frammento N-terminale (dal residuo 1 al residuo 34), che viene escreto a livello dei tubuli renali, e il frammento C-terminale (dal residuo 35 al residuo 84) con più lunga emivita - circa 20 minuti -, che viene filtrato dai glomeruli e può accumularsi nel sangue in caso di insufficienza renale. Il paratormone presente in circolo è una miscela eterogenea di frammenti ed è costituito per il 10-20% dal peptide nativo e per il restante 80-90% dai sui prodotti di degradazione.
L’azione del paratormone è mediata da un recettore glicoproteico di membrana di 80 kDa, appartenente alla superfamiglia delle proteine G, che riconosce la porzione N-terminale del paratormone ed agisce stimolando la produzione di cAMP e di inositolo trifosfato. Il paratormone agisce a livello renale dove incrementa il riassorbimento del calcio (vedi Par. 12.4) ed inibisce il riassorbimento del fosfato (vedi Par. 12.6), causando ipercalcemia ed ipofosfatemia; l’ormone inoltre inibisce lo scambio NA+/H+ e il riassorbimento del bicarbonato (vedi Par. 12.1), causando una lieve acidosi metabolica. L’azione a livello gastroenterico è mediata in gran parte dalla 1α,25-diidrossi-vitamina D, la cui sintesi è promossa dal paratormone (vedi Par. 10.9d). L’azione a livello osseo è complessa: ad alte concentrazioni, come quelle osservabili in caso di iperparatiroidismo primario o secondario, il paratormone favorisce il riassorbimento osseo mediato dagli osteoclasti, mentre quando l’aumento del paratormone è piccolo, specialmente se episodico, come nel caso del trattamento dell’osteoporosi mediante iniezioni giornaliere dell’ormone, il paratormone favorisce la formazione dell’osso mediata dagli osteoblasti.
10.9b. Proteina correlata al paratormone
La proteina correlata al paratormone (PTHrP, parathyroid hormone-related protein) è un ormone polipeptidico che è prodotto in diversi tessuti fetali, ma con il progredire dello sviluppo la sua sintesi viene sempre più a ridursi. La sua increzione aumenta nuovamente in diversi tipi di tumori maligni (in particolare nel cancro della mammella, della prostata e del polmone). Il PTHrP si lega al recettore del paratormone ed induce gran parte degli effetti metabolici propri del paratormone, compresa l’ipercalcemia. Regola la proliferazione e la mineralizzazione della cartilagine ed è necessario per un normale sviluppo scheletrico. Il gene del PTHrP codifica per tre polipeptidi diversi (PTHrP 1-139, PTHrP 1-141 e PTHrP 1-173) mediante un meccanismo di splicing alternativo del mRNA. Questi si legano a specifici recettori e producono effetti biologici distinti.
10.9c. Calcitonina
La calcitonina è un peptide di 32 aminoacidi increto dalle
cellule parafollicolari C della tiroide
![]() . L’increzione
dell’ormone è stimolata dall’ipercalcemia, mentre è inibita dall’ipocalcemia. Il
principale effetto della calcitonina è di inibire il riassorbimento osseo
mediato dagli osteoclasti. La calcitonina inibisce il riassorbimento renale del
fosfato, promuove l’escrezione di questo anione e, più debolmente, l’escrezione
del sodio e del calcio.
. L’increzione
dell’ormone è stimolata dall’ipercalcemia, mentre è inibita dall’ipocalcemia. Il
principale effetto della calcitonina è di inibire il riassorbimento osseo
mediato dagli osteoclasti. La calcitonina inibisce il riassorbimento renale del
fosfato, promuove l’escrezione di questo anione e, più debolmente, l’escrezione
del sodio e del calcio.
10.9d. Vitamina D
Con il termine "vitamina D" si indica un insieme di
pro-ormoni secosteroidei, analoghi al colesterolo ma con l’anello B aperto (vedi
Par. 3.3), i cui rappresentanti
principali sono l’ergocalciferolo (vitamina D2) e il colecalciferolo
(vitamina D3). La vitamina D2 si
differenzia dalla vitamina D3 per la presenza di un doppio legame fra
gli atomi di carbonio 22 e 23 e per un gruppo metile sul carbonio 24. La
vitamina D2 deriva dall’ergosterolo, uno steroide presente nelle
piante e nei funghi, ma assente nell’uomo. La vitamina D3 è presente
nel regno animale e si forma nella cute dell’uomo, principalmente nello strato
basale e spinoso, per fotolisi del 7-deidrocolesterolo (vedi Fig. 10.15,
![]() ). Per produrre una quantità adeguata di vitamina D3 è
necessario esporre almeno un paio di volte alla settimana la cute (viso,
braccia, o torace) ai raggi diretti del sole per 10-15 minuti durante la
stagione primaverile ed estiva nelle zone temperate o in un periodo
qualsiasi dell’anno ai tropici. Esposizioni prolungate al sole non portano ad una
sovrapproduzione di vitamina D3 in quanto la quota in eccesso viene
degradata subito dopo essere stata prodotta.
). Per produrre una quantità adeguata di vitamina D3 è
necessario esporre almeno un paio di volte alla settimana la cute (viso,
braccia, o torace) ai raggi diretti del sole per 10-15 minuti durante la
stagione primaverile ed estiva nelle zone temperate o in un periodo
qualsiasi dell’anno ai tropici. Esposizioni prolungate al sole non portano ad una
sovrapproduzione di vitamina D3 in quanto la quota in eccesso viene
degradata subito dopo essere stata prodotta.
Fig. 10.15. Sintesi e idrossilazione della vitamina D. Vitamina D2: ergocalciferolo; vitamina D3: colecalciferolo; 25(OH)D2: 25-idrossi-ergocalciferolo; 1α,25(OH)D2: 1α,25-diidrossi-ergocalciferolo; P450c25: 25-idrossilasi; P450c1: 25-idrossi-vitamina D 1-idrossilasi.
L’ergocalciferolo e il colecalciferolo subiscono una
idrossilazione in posizione 25, catalizzata da una monoossigenasi
mitocondriale (un citocromo P450) presente nel fegato, e
successivamente una idrossilazione in posizione 1a, catalizzata dalla 25-idrossi-vitamina
D 1-idrossilasi, un enzima presente nel rene
![]() che è attivato dal paratormone e dalla calcitonina
ed è inibito dal calcio e dal fosfato inorganico. La 25-idrossi-vitamina D e la
1α,25-diidrossi-vitamina D sono substrati dell’enzima
24-idrossilasi, una proteina presente in numerosi tessuti e coinvolta nel
processo di inattivazione catabolica della vitamina D. Tutti i derivati della
vitamina D sono liposolubili e trasportati nel sangue legati in gran parte
alla transcalciferina (group
component), una α-globulina
strutturalmente omologa all’albumina e all’α-fetoproteina
che è attivato dal paratormone e dalla calcitonina
ed è inibito dal calcio e dal fosfato inorganico. La 25-idrossi-vitamina D e la
1α,25-diidrossi-vitamina D sono substrati dell’enzima
24-idrossilasi, una proteina presente in numerosi tessuti e coinvolta nel
processo di inattivazione catabolica della vitamina D. Tutti i derivati della
vitamina D sono liposolubili e trasportati nel sangue legati in gran parte
alla transcalciferina (group
component), una α-globulina
strutturalmente omologa all’albumina e all’α-fetoproteina
![]() .
.
La 1α,25-diidrossi-vitamina D, che è prodotta dal rene e riversata nel torrente circolatorio, si comporta come un ormone, contribuendo alla regolazione della trascrizione genica in diversi distretti corporei ed intervenendo nei processi di proliferazione e differenziazione cellulare. La 1α,25-diidrossi-vitamina D inibisce la sintesi del paratormone da parte delle ghiandole paratiroidi e favorisce i processi di mineralizzazione delle ossa, incrementando l’assorbimento del calcio (vedi Par. 12.4) e dei fosfati (vedi Par. 12.6) a livello intestinale. La 1α,25-diidrossi-vitamina D favorisce inoltre il riassorbimento renale del calcio e del fosfato, ma la sua importanza a livello del rene è sicuramente minore rispetto a quella del paratormone (vedi Par. 10.9a).
10.9.1. Metodi di determinazione
Gli ormoni coinvolti nella regolazione del metabolismo
osseo sono generalmente dosati nella routine clinica mediante
metodi immunologici basati sulla competizione, rispetto ad un numero
limitato di siti anticorpali, del campione in esame con una preparazione
standard dell’analita, che è stato reso evidenziabile mediante
mararcatura con un elemento radioattivo, un fluoroforo o un enzima. Il
complesso antigene-anticorpo viene isolato mediante precipitazione in
presenza di carbone attivo o mediante l’utilizzo di un secondo anticorpo
insolubilizzato
![]() .
L’eccesso di materiale marcato viene quindi allontanato dall’aggregato
macromolecolare mediante lavaggio.
.
L’eccesso di materiale marcato viene quindi allontanato dall’aggregato
macromolecolare mediante lavaggio.
Per quanto riguarda il dosaggio del paratormone, l’interpretazione dei risultati varia a seconda del tipo di anticorpi adoperato. Gli anticorpi che si legano in prossimità della zona amino-terminale dell’ormone (N-assay) evidenziano sia la molecola intera sia il frammento N-terminale, entrambi a emivita molto breve, mentre gli anticorpi che legano la zona carbossi-terminale dell’ormone (C-assay) evidenziano essenzialmente il frammento C-terminale, a emivita più lunga. Bisogna inoltre tenere presente che il residuo amino-terminale, importante per l’azione biologica dell’ormone, non è generalmente riconosciuto dagli anticorpi presenti nelle confezioni designate per intact PTH assays.
Per valutare indirettamente l’attività del paratormone in vivo, è possibile dosare il cAMP eliminato con le urine (cAMP urinario). Questo comprende una quota di cAMP liberato a livello renale per azione del paratormone e una quota di derivazione plasmatica di diversa origine, che è stata filtrata dal glomerulo. Il livello plasmatico di paratormone correla bene con il "cAMP nefrogeno", che è definito dalla differenza fra il cAMP urinario e il cAMP di derivazione plasmatica, calcolabile dal suo rapporto di clearance rispetto alla creatinina. Il livello di cAMP nefrogeno aumenta in caso di iperparatiroidismo, deficit di vitamina D e ipercalcemia in pazienti affetti da cancro.
Per quanto riguarda la vitamina D, il saggio in presenza di anticorpi specifici dà luogo a risultati scarsamente attendibili quando viene utilizzato su campioni contenenti dei derivati sia dell’ergocalciferolo che del colecalciferolo in quanto entrambe queste classi di composti si possono legare agli anticorpi, ma con diversa affinità, rendendo pertanto difficile l’interpretazione del dato analitico. Poiché la transcalciferina ha una elevata affinità per i derivati della vitamina D, preparazioni parzialmente purificate di questa proteina possono essere utilizzate al posto degli anticorpi in questo tipo di saggi (Competitive Protein-Binding Assay, CPBA). I metodi che utilizzano la transcalciferina risentono in maggior misura della presenza di sostanze interferenti eventualmente presenti nel campione in esame.
Il metodo di riferimento per il dosaggio della 25-idrossi-vitamina D, utilizzato principalmente nei laboratori di ricerca, prevede la separazione cromatografica (mediante cromatografia liquida ad alta pressione, HPLC) degli analiti e la determinazione della loro concentrazione mediante uno spettrofotometro o uno spettrometro di massa. La concentrazione della 1α,25-diidrossi-vitamina D, nella frazione separata mediante cromatografia, viene determinata utilizzando metodi RIA che prevedono l’uso di anticorpi o di preparazioni di recettori intestinali di pulcino specifici per questo metabolita.
10.9.2. Preparazione del campione ed intervalli di riferimento
La determinazione del paratormone può essere eseguita su siero o plasma in EDTA; il paratormone è sottostimato nel plasma trattato con eparina. A fini diagnostici, è conveniente analizzare campioni prelevati al paziente in tre giorni differenti e determinare in essi anche il livello di calcio e di creatinina. I campioni devono essere conservati a -70°C per impedire che vengano degradati.
La determinazione della vitamina D e dei sui metaboliti può essere eseguita su siero o su plasma. La 25-idrossi-vitamina D è il più abbondante fra i vari metaboliti della vitamina D presenti in circolo ed è quello con maggiore emivita (1-2 settimane); la sua concentrazione ematica è un indice spesso utilizzato per valutare la sintesi della vitamina a livello cutaneo e/o la sua introduzione con la dieta. Deficit alimentari di vitamina D, rachitismo e osteomalacia sono associati a bassi livelli ematici di 25-idrossi-vitamina D (< 4 ng/mL), mentre soggetti intossicati con la vitamina D hanno alti livelli ematici di questo analita (>200 ng/mL). La 1α,25-diidrossi-vitamina D ha una emivita di circa 15 ore e concentrazioni ematiche circa 100 volte inferiori (vedi Tab. 10.III).
Tab. 10.III. Livelli sierici di derivati della vitamina D in soggetti adulti in una zona temperata durante il periodo estivo. La concentrazione sierica di ergocalciferolo (non riportata in tabella) dipende largamente dalla dieta.
|
ng/mL |
||
| colecalciferolo | 10 | |
| 25-idrossi-vitamina D | 25 | |
| 1α,25-diidrossi-vitamina D | 00.03 | |
| 24,25-diidrossi-vitamina D | 01 | |
| aggiornamento: 04/10/14 |