|
|
Costantino Salerno |
|
|
Appunti di Biochimica Clinica |
|
|
14. ANALISI GENETICHE
Sono definite "genetiche" quelle malattie riconducibili ad alterazioni del DNA nucleare o mitocondriale. Le malattie genetiche possono essere ereditarie se le alterazioni sono presenti in uno o entrambi i genitori, o se riguardano soltanto la cellula uovo o lo spermatozoo negli stadi che immediatamente precedono la formazione dello zigote. Le malattie genetiche sono acquisite quando sono dovute ad alterazioni del DNA verificatesi successivamente alla formazione dello zigote, come ad esempio può accadere in molti tipi di neoplasie.
Le malattie dovute ad alterazioni del DNA nucleare possono essere conseguenti a (1) mancanza, eccesso o riarrangiamento anomalo di uno o più cromosomi o parti di essi, se queste modificazioni sono tali da influenzare la struttura e la funzione di uno o più geni, (2) variazioni più o meno estese riguardanti un singolo gene, (3) alterazioni a carattere multifattoriale riferite a più geni e/o ad interazioni di questi con uno o più fattori esogeni o ambientali.
14.1. ANOMALIE CROMOSOMICHE
Le anomalie cromosomiche possono riguardare il numero (anomalie numeriche), la morfologia (anomalie strutturali) o l’espressione differenziale (imprinting) di uno o più cromosomi.
14.1.1. Anomalie numeriche
Con il termine "poliploidia" si intende la presenza di un multiplo intero, superiore a due, di un assetto cromosomico aploide normale (Fig. 14.1). Poiché il cariotipo normale dell’uomo è costituito da 46 cromosomi, ossia 22 coppie di cromosomi autosomici omologhi e una coppia di cromosomi sessuali (XX o XY), si possono in teoria osservare triploidie corrispondenti a 69 cromosomi, tetraploidie corrispondenti a 92 cromosomi, e così via. In pratica le poliploidie sono sempre letali e solo in pochissimi casi in un nato vivo si può riscontrare una triploidia vera.
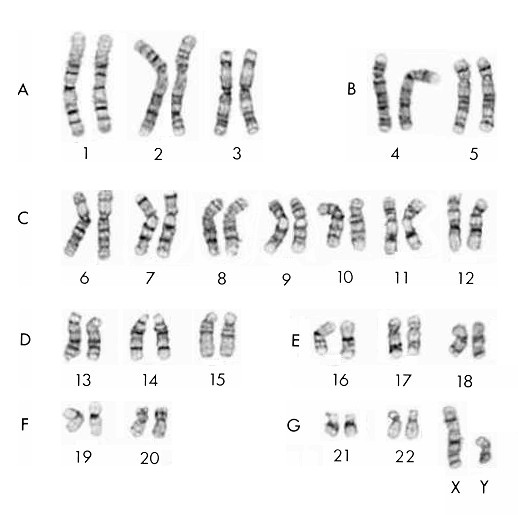
Fig. 14.1. Cariotipo maschile normale. I cromosomi sono classificati in sette gruppi (da A a G) in ordine decrescente di grandezza ed in base all’indice centromerico, ossia al rapporto tra la lunghezza del braccio minore e quella dell’intero cromosoma. Il cromosoma X rientra per dimensioni e caratteristiche morfologiche nel gruppo C, mentre il cromosoma Y è simile ai cromosomi del gruppo G. I singoli cromosomi possono essere riconosciuti attraverso tecniche di bandeggiatura. Queste tecniche consistono nell’aggiunta di opportuni coloranti (quinacrina, mostarda di quinacrina, arancio di acridina, etc.), in trattamenti di denaturazione e rinaturazione seguiti da colorazione con Giemsa, in trattamenti con enzimi proteolitici seguiti a colorazione o nell’uso di opportune sonde marcate attraverso metodiche di ibridazione in situ (Vedi Par. 14.6.6d).
Con il termine "aneuploidia" si definisce un assetto
cromosomico in cui il numero di cromosomi varia di una unità in
più o in meno rispetto al normale (si parla di monosomia quando
il soggetto ha 45 cromosomi e di trisomia quando ne ha 47). Le
monosomie hanno effetti fenotipici più gravi delle trisomie e
non si ritrovano praticamente mai monosomie degli autosomi
neanche tra gli aborti precoci. Una monosomia caratterizzata
dalla presenza di un solo cromosoma sessuale (il cromosoma X) è
riscontrabile nella sindrome di Turner
![]() . Le trisomie più
frequenti sono la sindrome di Klinefelter (1/800
maschi,
. Le trisomie più
frequenti sono la sindrome di Klinefelter (1/800
maschi,
![]() ),
la sindrome di Jacobs (1/1000 maschi,
),
la sindrome di Jacobs (1/1000 maschi,
![]() ), la sindrome di
Down (1/800 nati,
), la sindrome di
Down (1/800 nati, ![]() ), la sindrome di Edwards (1/6000 nati,
), la sindrome di Edwards (1/6000 nati,
![]() )
e la sindrome di Patau (1/10000 nati,
)
e la sindrome di Patau (1/10000 nati,
![]() ).
).
14.1.2. Anomalie strutturali
Le anomalie strutturali dei cromosomi possono essere
bilanciate, quando la quantità globale del materiale genetico è
immutata, ma è cambiata la disposizione dei geni lungo il
cromosoma
![]() ,
o sbilanciate quando la quantità globale del materiale genetico
è diversa dal normale.
,
o sbilanciate quando la quantità globale del materiale genetico
è diversa dal normale.
Le anomalie riguardanti un solo cromosoma comprendono le inversioni (dovute ad una rotazione di 180° di un segmento cromosomico compreso fra due rotture), le delezioni (o monosomie parziali, causate dalla perdita di un segmento di un cromosoma), le duplicazioni (o trisomie parziali, dovute al raddoppio di un segmento di un cromosoma), i cromosomi ad anello (quando si ha la circolarizzazione del cromosoma con perdita delle sue porzioni distali) e gli isocromosomi (o cromosomi simmetrici, costituiti da cromosomi con le due braccia di uguale lunghezza per traslocazione reciproca delle braccia lunghe e corte fra due cromosomi omologhi).
Le anomalie che interessano più cromosomi comprendono le inserzioni (dovute all’introduzione di un segmento cromosomico all’interno di un secondo cromosoma), le traslocazioni reciproche (dovute allo scambio reciproco di segmenti tra cromosomi non omologhi) e le traslocazioni Robertsoniane (in cui due cromosomi acrocentrici si uniscono per il centromero dopo aver perso il braccio corto).
14.1.3. Anomalie dell’imprinting genomico
L’imprinting genomico è la condizione che si verifica normalmente quando l’espressione fenotipica di determinati geni o cromosomi dipende dal sesso del genitore da cui essi provengono. Tale possibilità dipende dai meccanismi molecolari che regolano l’espressione genica, fra cui, in particolare, la metilazione differenziale del DNA. Questi meccanismi possono lasciare attivi (cioè codificanti) in uno o più loci solo i geni di origine paterna (imprinting materno), mentre in altri loci solo quelli di origine materna (imprinting paterno), realizzando in entrambi i casi una condizione di disomia strutturale con monosomia funzionale.
Numerose osservazioni hanno confermato l’esistenza di tale fenomeno sia negli animali da esperimento che nell’uomo. Esperimenti di trapianto di pronuclei hanno dimostrato che gli embrioni con patrimonio diploide interamente paterno (androgenici) o interamente materno (partenogenici) si sviluppano in modo diverso. Nelle triploidie umane si è visto che i patrimoni aploidi paterno e materno sono entrambi necessari, ma complementari allo sviluppo dell’embrione e della placenta. Infatti, in aggiunta alle specifiche malformazioni fetali, i triploidi diandrici (con due cromosomi di origine paterna ed uno di origine materna) mostrano solitamente una iperplasia del trofoblasto, mentre nei triploidi diaginici (con due cromosomi di origine materna ed uno di origine paterna) si osserva generalmente una placenta fortemente sottosviluppata. La dimostrazione più evidente del fenomeno dell’imprinting è fornita dalle particolari espressioni fenotipiche di alcune alterazioni cromosomiche non sempre visibili citogeneticamente, ma sempre dimostrabili mediante l’uso di opportune sonde (vedi Par. 14.6.6c). Le alterazioni più comuni sono a carico del cromosoma 15 e danno luogo alle sindromi di Prader-Willi e di Angelman.
La sindrome di Prader-Willi
![]() si
può verificare quando si ha una delezione della regione 15q11-q13
sul cromosoma di origine paterna, dove è contenuta, per imprinting
genomico materno, la sola copia funzionalmente attiva dei geni
critici, in modo da realizzare una condizione di monosomia
strutturale con nullisomia funzionale. Un’altra causa può
essere una condizione di disomia uniparentale materna del
cromosoma 15 che porta invece ad una disomia strutturale con
nullisomia funzionale
si
può verificare quando si ha una delezione della regione 15q11-q13
sul cromosoma di origine paterna, dove è contenuta, per imprinting
genomico materno, la sola copia funzionalmente attiva dei geni
critici, in modo da realizzare una condizione di monosomia
strutturale con nullisomia funzionale. Un’altra causa può
essere una condizione di disomia uniparentale materna del
cromosoma 15 che porta invece ad una disomia strutturale con
nullisomia funzionale ![]() .
.
Nella sindrome di Angelman
![]() si
ha una situazione simmetricamente inversa alla sindrome di Prader-Willi.
Infatti, nel 75-85% dei casi si ha una delezione della regione 15q11-q13
del cromosoma di provenienza materna (contenente la copia
funzionalmente attiva dei geni implicati nella sindrome di
Angelman), mentre in altri casi è spesso dimostrabile una
condizione di disomia uniparentale paterna. In altri casi ancora
è stata descritta una mutazione da cause sconosciute dell’allele
di origine materna con un suo conseguente difetto di espressione.
si
ha una situazione simmetricamente inversa alla sindrome di Prader-Willi.
Infatti, nel 75-85% dei casi si ha una delezione della regione 15q11-q13
del cromosoma di provenienza materna (contenente la copia
funzionalmente attiva dei geni implicati nella sindrome di
Angelman), mentre in altri casi è spesso dimostrabile una
condizione di disomia uniparentale paterna. In altri casi ancora
è stata descritta una mutazione da cause sconosciute dell’allele
di origine materna con un suo conseguente difetto di espressione.
Una situazione più complessa è riscontrabile in caso di errore congenito sulla subunità α (codificata nella regione q13 del cromosoma 20) della proteina Gs coinvolta nel sistema di trasduzione del segnale per il paratormone. Il deficit determina un quadro dismorfico caratteristico (osteodistrofia ereditaria di Albright). A causa di un imprinting paterno sul gene della subunità α nelle cellule dei tubuli prossimali renali, la malattia è associata ad una resistenza al paratormone, quando il difetto è ereditato dalla madre (pseudoipoparatiroidismo tipo Ia; vedi Par. 12.4), mentre è limitata al solo quadro dismorfico con una normale risposta renale all’ormone, quando il difetto è ereditato dal padre (pseudo-pseudoipoparatiroidismo).
14.1.4. Sindromi da geni contigui
Le microdelezioni possono essere causa delle cosiddette
sindromi da geni contigui (Contiguous Gene Syndromes, CGS),
che sono caratterizzate da anomalie cromosomiche che determinano
una modificazione quantitativa (in difetto o in eccesso) della
dose di una serie di geni funzionalmente non correlati, ma
situati in una determinata regione cromosomica in immediata
prossimità l’uno dell’altro. Diverse malattie
genetiche sono classificabili in questo gruppo; fra queste
ricordiamo le sindromi di Prader-Willi e di Angelman (già
descritte nel Par.14.1.3) nonché le
sindromi di Beckwith-Wiedemann
![]() , Kallmann-de
Morsier
, Kallmann-de
Morsier
![]() , Di George
, Di George
![]() , Smith-Magenis
, Smith-Magenis
![]() e Williams-Beuren
e Williams-Beuren ![]() .
.
14.2. DIFETTI RIGUARDANTI SINGOLI GENI
I difetti riguardanti singoli geni comprendono le delezioni intrageniche (caratterizzate dalla perdita di un tratto del gene), le inserzioni (consistenti nell’introduzione di materiale genetico estraneo all’interno del gene), le duplicazioni (dovute ad un allungamento della catena per la duplicazione di un tratto relativamente ampio del gene), le espansioni (dovute alla ripetizione multipla di un singolo codone) e le mutazioni puntiformi (costituite da semplici sostituzioni di un nucleotide con un altro, senza alterare il numero totale dei nucleotidi nel gene). Per quanto riguarda il meccanismo di trasmissione, le malattie ereditarie possono essere autosomiche dominanti, autosomiche recessive, legate al cromosoma X o dipendenti da mutazioni dinamiche associate a trinucleotidi instabili.
14.2.1. Malattie autosomiche dominanti e recessive
I
caratteri autosomici possono essere dominanti o recessivi. Un
allele codificante per un tratto dominante esprime il suo
fenotipo nello stato eterozigote (cioè è sufficiente una sola
copia dell’allele perché si manifesti il suo effetto),
mentre un allele per un tratto recessivo manifesta il suo
fenotipo solo in caso di omozigosi. Se in un eterozigote vengono
espressi entrambi gli alleli ed ognuno di essi produce indipendentemente
il suo fenotipo, si parla di ereditarietà codominante. L’ereditarietà
codominante si osserva più chiaramente nei fenotipi studiati mediante test
biochimici o immunologici, come nel caso dei test per i gruppi
sanguigni
![]() .
.
Si ritiene che nella specie umana possano insorgere diverse migliaia di differenti malattie ereditarie monogeniche. Le malattie dominanti si associano spesso a geni che codificano per proteine dotate di funzioni strutturali, di trasporto o di recettore, mentre i geni che codificano per enzimi sono spesso associati a malattie recessive in quanto l’attività di ciascun enzima è il più delle volte considerevolmente in esubero rispetto al fabbisogno metabolico.
Sulla base dei rapporti di segregazione dettati dalla
genetica mendeliana, è logico attendersi che un carattere dominante sia
trasmesso verticalmente dal genitore affetto al 50% dei nati e in eguale misura
nei maschi e nelle femmine. I caratteri autosomici dominanti, quando favoriscono
lo sviluppo di patologie gravi, si manifestano generalmente solo durante o dopo
l’età riproduttiva e questo spiega l’alta frequenza di soggetti affetti da
condizioni altamente invalidanti in alcuni ceppi familiari (esempio la
neurofibromatosi
![]() , il rene
policistico
, il rene
policistico
![]() , la
sindrome di Lynch
, la
sindrome di Lynch
![]() , etc.).
, etc.).
Una malattia autosomica recessiva ha il 25% di probabilità di manifestarsi indipendentemente dal sesso nella progenie nata da un incrocio tra due portatori sani che, se la malattia è molto rara, spesso risultano essere consanguinei. Nel caso di caratteri autosomici recessivi non associati a fenotipi invalidanti sono tuttavia di comune riscontro anche gli incroci tra soggetti affetti dal difetto ereditario e soggetti normali così come gli incroci tra soggetti affetti dal difetto ereditario e portatori sani. Nel primo caso tutta la progenie sarà costituita da portatori fenotipicamente normali, mentre nel secondo caso il 50% della progenie risulterà fenotipicamente alterata.
Anche quando il gene si manifesta in tutti gli eterozigoti,
la malattia può presentarsi con diversi gradi di severità (espressività
variabile). In altri casi ancora (come ad esempio nella ipercolesterolemia
familiare
![]() ) si
osserva una minore gravità dei sintomi nell’eterozigote rispetto all’omozigote.
Queste ultime malattie si adattano male alla definizione mendeliana sia di
ereditarietà dominante, dove l’eterozigote deve essere simile all’omozigote, sia
di ereditarietà recessiva, dove l’eterozigote non differisce fenotipicamente dal
soggetto privo di anomalie genetiche.
) si
osserva una minore gravità dei sintomi nell’eterozigote rispetto all’omozigote.
Queste ultime malattie si adattano male alla definizione mendeliana sia di
ereditarietà dominante, dove l’eterozigote deve essere simile all’omozigote, sia
di ereditarietà recessiva, dove l’eterozigote non differisce fenotipicamente dal
soggetto privo di anomalie genetiche.
14.2.2. Malattie legate al cromosoma X
Le malattie dominanti legate al cromosoma X colpiscono sia i maschi che le
femmine. Le forme recessive (molto più frequenti) si manifestano unicamente nei
maschi. Le femmine sono generalmente portatrici sane dell’alterazione
genetica recessiva che solitamente si esprime solo in caso di omozigosi. Poiché
in ogni cellula solo un cromosoma X è attivo, la malattia può tuttavia
manifestarsi anche nell’eterozigote se i cromosomi X sono inattivati durante
l’embriogenesi femminile in modo tale da essere espressi nella forma mutante in
tessuti critici (ad esempio nel fegato nel caso di deficit di ornitina
transcarbamilasi
![]() ).
).
Una qualsiasi mutazione legata al cromosoma X si distribuisce nella
popolazione per i 2/3 nelle femmine, quasi sempre eterozigoti, e per 1/3 nei
maschi, emizigoti. Se la mutazione è geneticamente incompatibile con la
riproduzione, allora ad ogni generazione un terzo dei cromosomi X che portano la
mutazione va perso e ciò porta velocemente alla scomparsa della malattia nella
popolazione. Ad esempio, nel caso dell’emofilia A
![]() , una
malattia recessiva legata al cromosoma X che è stata analizzata da più tempo,
ciò che si osserva più spesso nello studio degli alberi genealogici è un solo
gruppo di fratelli affetti o un solo paziente in una famiglia per il resto sana
in quanto la ridotta capacità riproduttiva dei soggetti affetti dalla malattia
porta di norma alla eliminazione della maggior parte dei geni emofilici in poche
generazioni.
, una
malattia recessiva legata al cromosoma X che è stata analizzata da più tempo,
ciò che si osserva più spesso nello studio degli alberi genealogici è un solo
gruppo di fratelli affetti o un solo paziente in una famiglia per il resto sana
in quanto la ridotta capacità riproduttiva dei soggetti affetti dalla malattia
porta di norma alla eliminazione della maggior parte dei geni emofilici in poche
generazioni.
14.2.3. Mutazioni dinamiche associate a trinucleotidi instabili
Le malattie da trinucleotidi instabili sono forme ereditarie
dovute all’aumento del numero di triplette in sequenze CAG, CTG, CGG, o GAA reiterate, presenti in regioni esoniche, introniche o
regolative dei rispettivi geni. Queste sequenze sono normalmente
polimorfiche ![]() ed hanno una bassa frequenza di mutazione
del numero di ripetizioni. Tuttavia se, per ragioni non ancora
completamente note
ed hanno una bassa frequenza di mutazione
del numero di ripetizioni. Tuttavia se, per ragioni non ancora
completamente note ![]() , le ripetizioni superano una certa
lunghezza (generalmente intorno alle 30-40 triplette), le
sequenze diventano instabili, cioè aumenta in modo cospicuo la
probabilità che il numero di copie si espanda ulteriormente o
comunque si modifichi nella trasmissione alla generazione
successiva. L’instabilità del numero di copie negli alleli
espansi si verifica nella divisione sia di cellule germinali che
di cellule somatiche. Esiste perciò una eterogeneità del numero
di triplette degli alleli espansi sia tra genitori e figli che
tra le diverse cellule di uno stesso individuo
, le ripetizioni superano una certa
lunghezza (generalmente intorno alle 30-40 triplette), le
sequenze diventano instabili, cioè aumenta in modo cospicuo la
probabilità che il numero di copie si espanda ulteriormente o
comunque si modifichi nella trasmissione alla generazione
successiva. L’instabilità del numero di copie negli alleli
espansi si verifica nella divisione sia di cellule germinali che
di cellule somatiche. Esiste perciò una eterogeneità del numero
di triplette degli alleli espansi sia tra genitori e figli che
tra le diverse cellule di uno stesso individuo
![]() .
.
I processi patologici legati all’espansione della sequenza ripetuta si manifestano quando il numero di triplette supera una certa soglia, che non coincide necessariamente con quella per l’instabilità della sequenza stessa. Inoltre le malattie da trinucleotidi instabili sono caratterizzate da un aumento di penetranza della malattia stessa nel corso delle generazioni (paradosso di Sherman) e da una progressiva anticipazione dell’età di insorgenza e della gravità dei sintomi. Le malattie da trinucleotidi instabili sono classificate in due gruppi, a seconda che l’allungamento della sequenza avvenga in una porzione codificante o non codificante del gene interessato (Tab.I).
|
|
||||
|
malattia |
cromosoma |
prodotto genico |
trinucleotidi |
n. copie |
|
|
||||
|
Gruppo I |
|
|
|
|
|
4p16 |
huntingtina |
CAG |
36-121 |
|
|
6p23 |
atassia-1 |
CAG |
41-81 |
|
|
12q24-24 |
atassia-2 |
CAG |
32-77 |
|
|
14q24-32 |
atassia-3 |
CAG |
56-85 |
|
|
19p13 |
sub. a1A calcio |
CAG |
20-30 |
|
|
3p14-21 |
atassina-7 |
CAG |
37-200 |
|
|
12p12 |
atrofina |
CAG |
49-88 |
|
|
Xq12 |
rec. androgeni |
CAG |
36-62 |
|
|
Gruppo II |
|
|
|
|
|
19q13 |
miotonina chinasi |
CTG |
50-1000 |
|
|
Xq27-28 |
proteina FMR |
CGG |
200-1000 |
|
|
9q13 |
fratassina |
GAA |
120-1000 |
|
|
|
||||
Il primo gruppo è caratterizzato dall’allungamento di sequenze (CAG)n che vengono espresse a livello proteico come allungamenti dei tratti di poliglutamina normalmente presenti nella proteina codificata. Questo allungamento conferirebbe alla proteina nuove caratteristiche nocive (minore solubilità, possibilità di dar luogo ad aggregati insolubili endo- e perinucleari di poliglutamine), senza sottrarre ad essa le funzioni indispensabili, come è dimostrato dal fatto che i sintomi tipici della malattia sono assenti negli individui affetti da una delezione del gene interessato. In questo gruppo di malattie da trinucleotidi instabili, la variazione intergenerazionale del numero di triplette è maggiore e più frequente se la sequenza espansa è trasmessa dal padre. I sintomi sono caratterizzati da una progressiva degenerazione che colpisce specifiche zone cerebrali a seconda della malattia.
Il secondo gruppo è caratterizzato da espansioni di sequenze (CTG)n, (GAA)n o (CGG)n in regioni non codificanti dei rispettivi geni. L’espansione delle sequenze trinucleotidiche è molto più ampia rispetto al primo gruppo di malattie, raggiungendo fino un migliaio di copie. Il danno sarebbe dovuto ad una interferenza con i processi di trascrizione o di maturazione dell’mRNA. I maggiori aumenti del numero di ripetizioni avvengono quando la sequenza espansa è trasmessa dalla madre. Clinicamente, le malattie di questo gruppo sono caratterizzate da un impegno multisistemico.
14.2.4. Errori di "splicing"
I geni sono formati, di solito, da diversi tratti relativamente
brevi di DNA codificante (esoni)
intervallati da tratti generalmente lunghi di DNA non codificante (introni). I
tratti non codificanti, che sono presenti nel trascritto primario di mRNA, vengono rimossi e gli esoni sono riuniti a
formare una unica sequenza contigua (splicing). Nel caso dei trascritti primari
di mRNA nucleare, lo splicing è catalizzato dal spliceosoma, che è
formato da un complesso di cinque ribonucleoproteine nucleari (snRNP, small nuclear
ribonucleoprotein) e da numerose proteine non-snRNP, che hanno una
distribuzione specifica nei singoli tessuti. La sequenza di reazioni prevede l’attacco nucleofilo
da parte del gruppo 2’-OH di un particolare residuo adenilico, posto all’interno dell’introne, sul sito di splicing 5’
dell’introne stesso (splice donor
site) a formare una struttura ad anello; il gruppo 3’-OH dell’esone sul
versante
5’, che si è liberato in tale modo, può così agire da secondo agente nucleofilo
in una reazione analoga all’estremità 3’ dell’introne (splice acceptor site),
che porta al congiungimento degli esoni ed alla liberazione di un introne a
forma di laccio. Processi di
splicing alternativi, attraverso i quali un singolo trascritto primario
può dare luogo a differenti tipi di mRNA maturi, portano alla formazione di
isoforme proteiche con funzioni diverse, spesso contrastanti. Splicing
aberranti possono essere alla base di numerose malattie genetiche, come ad esempio la
β-talassemia (vedi
Par. 7.2.1b), la fibrosi cistica
![]() e
numerosi tipi di
cancro.
e
numerosi tipi di
cancro.
Un esempio di malattia genetica, dovuta ad un difetto primario di splicing, è l’atrofia spinale muscolare, che è caratterizzata da un errore nella proteina SMN (survival motor neuron protein). Questa proteina forma dei complessi con le ribonucleoproteine snRNP o con la proteina SIP (survival motor neuron-interacting protein) e, di conseguenza, un suo deficit impedisce ai motoneuroni di produrre mRNA che codifica per le proteine necessarie allo sviluppo e al funzionamento delle cellule.
Un esempio di splicing alternativo è osservabile nei pazienti affetti da disautonomia familiare (sindrome di Riley-Day). Questa malattia è caratterizzata da una mutazione sul splice donor site del gene IKBKAP (inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase complex-associated protein), che porta alla formazione di una proteina troncata nelle cellule del tessuto nervoso, ma permette ugualmente di sintetizzare proteine con una struttura normale in altre cellule (linfoblasti, fibroblasti).
14.2.5. Mutazioni silenti
Il trasferimento dell’informazione genetica dalla catena nucleotidica del mRNA alla catena polipeptidica, che verrà a costituire la proteina codificata, si realizza attraverso l’utilizzo di un codice, che mette in relazione ciascuna tripletta di basi (codone) con un aminoacido. Questo codice genetico ha la caratteristica di essere ridondante in quanto permette di costruire 64 (= 43) codoni diversi, quando per una corretta sintesi proteica sarebbe sufficiente indicare solo 20 aminoacidi più un segnale di arresto. A causa di questa ridondanza, per quasi tutti gli aminoacidi e per il segnale di arresto è presente più di un codone specifico. Per esempio, tutti i codoni che iniziano con GG (GGA, GGC, GGG, GGU) sono tradotti in glicina e, in questo senso, questi codoni sono da considerarsi "sinonimi" fra loro. Un elenco completo dei codoni sinonimi è riportato in Tab. 14.II.
Una mutazione puntiforme lungo la catena del DNA può portare alla sintesi di un mRNA che include o manca di un segnale di arresto dove questo dovrebbe essere presente, contiene un codone sbagliato (specifico per un diverso aminoacido) oppure contiene un codone sinonimo. Mentre nei primi casi la mutazione ha come conseguenza la sintesi di una proteina più corta o più lunga del normale o la sostituzione di un aminoacido nella catena polipeptidica, nell’ultimo caso la mutazione non dovrebbe comportare alcuna alterazione nella sequenza aminoacidica della proteina codificata. Quest’ultimo tipo di mutazione viene pertanto definito "silente".
| UUU Fenilalanina | UCU Serina | UAU Tirosina | UGU Cisteina |
| UUC Fenilalanina | UCC Serina | UAC Tirosina | UGC Cisteina |
| UUA Leucina | UCA Serina | UAA Stop | UGA Stop |
| UUG Leucina | UCG Serina | UAG Stop | UGG Triptofano |
| CUU Leucina | CCU Prolina | CAU Istidina | CGU Arginina |
| CUC Leucina | CCC Prolina | CAC Istidina | CGC Arginina |
| CUA Leucina | CCA Prolina | CAA Glutammina | CGA Arginina |
| CUG Leucina | CCG Prolina | CAG Glutammina | CGG Arginina |
| AUU Isoleucina | ACU Treonina | AAU Asparagina | AGU Serina |
| AUC Isoleucina | ACC Treonina | AAC Asparagina | AGC Serina |
| AUA Isoleucina | ACA Treonina | AAA Lisina | AGA Arginina |
| AUG Metionina | ACG Treonina | AAG Lisina | AGG Arginina |
| GUU Valina | GCU Alanina | GAU Aspartato | GGU Glicina |
| GUC Valina | GCC Alanina | GAC Aspartato | GGC Glicina |
| GUA Valina | GCA Alanina | GAA Glutammato | GGA Glicina |
| GUG Valina | GCG Alanina | GAG Glutammato | GGG Glicina |
In linea di principio, tutte le mutazioni che non esercitano effetti sull’organismo dovrebbero essere insensibili al processo di selezione naturale, che premia e preserva le variazioni che risultano benefiche. Si è scoperto tuttavia che in molti casi i segmenti di DNA, che contengono siti silenti, non presentano grandi variazioni nella popolazione o sono soggetti ad evoluzione lenta rispetto ad altre zone del DNA; ciò ha portato ad ipotizzare che anche le mutazioni nei siti silenti potrebbero risultare dannose ed essere pertanto soggette a un processo di eliminazione selettiva. Sono stati ipotizzati diversi meccanismi per spiegare la patogenesi di malattie legate a mutazioni silenti, tutti correlati a ruoli accessori che possono essere di competenza della base soggetta a mutazione.
Una mutazione silente in una
zona dell’esone contenente un esonic splicing enhancer (ESE) può
rendere invisibile questa sequenza ai meccanismi di splicing (vedi
Par. 14.2.4) facendo sì che un intero esone sia lasciato
fuori dal mRNA maturo. L’ESE è costituito da una breve sequenza nucleotidica
all’interno dell’esone, capace di interagire con le proteine regolatrici dello
splicing e dirigere gli spliceosomi a legarsi alle due estremità
dell’introne operandone la sua scissione. Un disturbo dello
splicing del gene CFTR (Cystic
Fibrosis Transmembrane conductance Regulator) a seguito di una mutazione silente
può contribuire, ad esempio,
alla fibrosi cistica o ad altri disturbi ad essa correlati
![]() .
.
Un deficit del recettore D2 della dopamina, che è riscontrablile in alcune forme di schizofrenia, è dovuto ad una mutazione silente che interferisce con il ripiegamento a forcina (stem-loop) del mRNA maturo e rende più rapida del normale la sua degradazione. Al contrario, nel caso del deficit di catecol-O-metiltransferase, un enzima che modula il ruolo delle catecolamine nella funzione cognitiva e nella capacità di sopportare il dolore, una mutazione silente intensifica il ripiegamento del mRNA maturo, che diviene più difficile da srotolare, ostacolando il processo di traduzione durante la sintesi proteica.
Un altro esempio, in cui una mutazione silente può influire su una proteina, riguarda il gene della resistenza multipla ai farmaci, che codifica per una pompa cellulare che, nelle cellule tumorali, contribuisce ad espellere i chemioterapici, rendendo le cellule resistenti alle terapie. In questo caso si ipotizza che il codone sinonimo dia origine ad un rallentamento del processo di traduzione ribosomiale, consentendo alla proteina nascente di ripiegarsi ed adottare una struttura insolita.
14.3. ALTERAZIONI A CARATTERE MULTIFATTORIALE
Le malattie multifattoriali o poligeniche sono quelle malattie ereditarie non riconducibili ad una patologia di tipo monogenico con ereditarietà mendeliana. I rapporti di segregazione osservati per molte di queste malattie sembrano adattarsi ad un modello multifattoriale a soglia che presuppone l’esistenza di un numero relativamente elevato di geni sfavorevoli, non necessariamente patologici, distribuiti a caso nella popolazione, che agirebbero in combinazione con fattori ambientali negativi, presenti nella vita fetale o dopo la nascita. Malattie come l’aterosclerosi, l’ipertensione, il diabete, le affezioni autoimmuni, diversi processi di natura neuropsichiatrica e cardiovascolare, molte allergie e alcuni tumori rappresentano esempi di patologie multifattoriali.
Le anomalie ereditarie alla base delle malattie
multifattoriali mostrano generalmente una correlazione inversa
tra prevalenza nella popolazione e penetranza del difetto
genetico. Poiché le mutazioni con penetranza elevata (ovvero
quelle che predispongono fortemente a una malattia) sono anche le
meno diffuse, lo screening per queste mutazioni ha un
senso soltanto se effettuato all’interno di famiglie in cui
la malattia è frequente. Infatti, sottoporre a screening
una popolazione per una mutazione poco diffusa significa
analizzare migliaia di individui per trovare pochissimi casi
![]() ,
con un rapporto fra costi e benefici decisamente sfavorevole
anche in considerazione dell’elevato rischio di trovare dei
soggetti falsi positivi, per i quali comunque dovrebbero essere
intrapresi accertamenti e cure non necessarie. Per geni a bassa
penetranza ed alta prevalenza, sarebbe sufficiente esaminare un
numero minore di pazienti per trovare le mutazioni cercate. In
questi casi però l’anomalia del DNA è soltanto uno dei
fattori che possono favorire lo sviluppo della malattia. Essere
portatori della mutazione non significa necessariamente
ammalarsi, né è chiaro cosa fare se si ottiene un risultato
positivo, in quanto per molte malattie che hanno una componente
genetica non esistono cure efficaci.
,
con un rapporto fra costi e benefici decisamente sfavorevole
anche in considerazione dell’elevato rischio di trovare dei
soggetti falsi positivi, per i quali comunque dovrebbero essere
intrapresi accertamenti e cure non necessarie. Per geni a bassa
penetranza ed alta prevalenza, sarebbe sufficiente esaminare un
numero minore di pazienti per trovare le mutazioni cercate. In
questi casi però l’anomalia del DNA è soltanto uno dei
fattori che possono favorire lo sviluppo della malattia. Essere
portatori della mutazione non significa necessariamente
ammalarsi, né è chiaro cosa fare se si ottiene un risultato
positivo, in quanto per molte malattie che hanno una componente
genetica non esistono cure efficaci.
14.4. MALATTIE MITOCONDRIALI
Il DNA mitocondriale umano, presente in 2-10 copie in ogni mitocondrio, è una molecola circolare costituita da 16569 paia di basi e codificante per 2 RNA ribosomiali (12 S e 16 S), 22 RNA transfer e 13 RNA messaggeri. I 13 peptidi codificati dal DNA mitocondriale sono costituenti della catena respiratoria. Le principali sindromi legate ad alterazioni del DNA mitocondriale sono riportate in Tab. 14.III.
Tab. 14.III. Principali difetti genetici del DNA mitocondriale |
|
| s. di Kearns-Sayre (KSS) | oftalmoplegia progressiva esterna, degenerazione pigmentaria della retina, insorgenza giovanile |
| s. di Leber (LHON) | neuropatia ottica ereditaria |
| s. di Leigh | encefalomiopatia necrotizzante subacuta |
| s. di Pearson | anemia sideroblastica, deficit del pancreas esocrino |
| MELAS | encefalomiopatia mitocondriale, acidosi lattica, emiparesi ricorrenti |
| MERRF | epilessia mioclonica, miopatia con ragged-red fibers |
| MIDD | diabete mellito, sordità |
| NARP | debolezza muscolare neurogena, atassia, retinite pigmentosa |
| PEO | oftalmoplegia esterna progressiva |
| MNGIE | encefalomiopatia, alterazione della motilità intestinale |
I difetti riguardanti il DNA mitocondriale sono trasmessi solo per via materna e colpiscono sia i maschi che le femmine, dal momento che tutti i mitocondri presenti nello zigote sono derivati dalla cellula uovo. Nella maggior parte dei casi, le cellule sono eteroplasmiche, cioè contengono sia mitocondri con DNA normale che mitocondri con DNA alterato. Poiché la distribuzione dei mitocondri nelle cellule figlie è stocastica, la percentuale di mitocondri con DNA alterato è molto variabile. Questo processo di segregazione mitotica, se si verifica nella divisione meiotica dell’uovo, determina una variabilità nelle manifestazioni fenotipiche della malattia nei figli della stessa madre portatrice, mentre, se si verifica durante le divisioni mitotiche delle cellule somatiche dell’embrione, dà luogo ad un diverso grado di deficit funzionale nei vari organi. Inoltre, a causa della segregazione mitotica, il permanere del difetto genetico tende ad essere limitato dalla morte della cellula figlia, se questa riceve una quantità di DNA alterato incompatibile con il suo metabolismo. Questo fenomeno è riscontrabile soprattutto nei tessuti in attiva moltiplicazione e, in vitro, nelle colture cellulari.
Un’altra importante conseguenza della eteroplasmia cellulare è rappresentata dall’effetto soglia. La cellula mostra infatti sofferenza per l’alterazione del DNA mitocondriale quando la quantità del DNA normale scende sotto un valore critico (sotto il 10-20% nei pazienti affetti da MERRF). Va tuttavia osservato che il DNA mitocondriale ha un tasso di mutazione di un ordine di grandezza più elevato rispetto al DNA nucleare per l’assenza di sistemi riparativi. Nei pazienti con mitocondriopatie, è perciò possibile che il DNA normale sia superiore alla soglia critica nei primi anni di vita e che la sintomatologia si manifesti più tardi. L’accumulo di alterazioni del DNA mitocondriale durante la vita porterebbe inoltre ad un progressivo deficit funzionale dei mitocondri nei tessuti costituiti da cellule che hanno perduto la capacità mitotica (come ad esempio le cellule muscolari), anche a causa della produzione di grandi quantità di radicali liberi dell’ossigeno nei mitocondri stessi.
Una forma particolare di malattia mitocondriale è rappresentata dalla encefalopatia neurogastrointestinale mitocondriale (MNGIE). A differenza delle altre malattie mitocondriali, questa malattia si trasmette come un carattere autosomico in quanto la causa primaria risiede in un deficit di timidina fosforilasi, dovuto a sua volta ad una mutazione puntiforme del gene presente nel braccio lungo del cromosoma 22. Nella MNGIE, l’alterazione del DNA mitocondriale non è perciò primitiva, ma secondaria ad uno squilibrato rapporto tra nucleotidi purinici e pirimidinici che porta ad un incremento degli errori nella duplicazione del genoma dei mitocondri che, come si è detto, sono costituzionalmente privi di meccanismi di riparo.
Le malattie mitocondriali presentano una sintomatologia che dipende dall’interessamento, nell’ordine, del sistema nervoso centrale, del muscolo scheletrico, del cuore, dei reni e del fegato. Questa gerarchia dipende dalla diversa efficienza del metabolismo mitocondriale nei vari tessuti. Se la malattia compare precocemente ed è multisistemica, si osserva frequentemente un rallentamento delle crescita corporea. Il quadro ematochimico è frequentemente rappresentato da acidosi lattica con aumento anche dell’acido piruvico e dell’alanina.
14.5. NEOPLASIE
Le principali forme di alterazioni geniche che possono portare a trasformazioni neoplastiche sono quelle che producono oncogeni (a partire da protooncogeni cellulari) o che inducono modificazioni della normale funzionalità di geni soppressori e/o di antioncogeni. In generale, gli oncogeni sono prodotti da mutazioni geniche dominanti, mentre nel caso dei geni oncosoppressori la mutazione è recessiva.
Lo sviluppo di un tumore si compie a seguito dell’accumulo di un numero di mutazioni (variabile a seconda del tipo cellulare) che insorgono in una data sequenza nella progenie di una singola cellula. La predisposizione ereditaria allo sviluppo di tumori è quella condizione in cui un particolare difetto genetico ricorrente in un albero genealogico determina o una riduzione del numero di mutazioni somatiche richieste per il compimento del processo canceroso o un aumento della frequenza delle mutazioni stesse. Sia nel caso di predisposizione ereditaria che nel caso di processi sporadici, l’insorgenza di mutazioni è facilitata dalla instabilità genetica, cioè dalla perdita di fedeltà nei processi deputati alla replicazione, riparazione e segregazione del genoma durante il ciclo cellulare.
Una causa di instabilità è la presenza nel genoma di brevi
sequenze ripetitive, dette microsatellitari, che possono causare
un elevato numero di errori (slippages) durante l’azione
della DNA-polimerasi e che necessitano dell’attivazione di
efficienti meccanismi di riparazione. Si ritiene che l’instabilità
dei microsatelliti nel 13% dei cancri ereditari non poliposici
del colon (sindrome di Lynch
![]() )
sia dovuta ad una alterazione di questi meccanismi di
riparazione.
)
sia dovuta ad una alterazione di questi meccanismi di
riparazione.
Un’altra causa di instabilità è l’alterazione di uno dei meccanismi di controllo che assicurano l’ordine degli eventi nel ciclo cellulare, integrando la riparazione e la replicazione del DNA ed arrestando il ciclo stesso durante il differenziamento, la senescenza, la morte cellulare programmata (apoptosi) o quando è stata compromessa l’integrità del genoma. Ad esempio, una mutazione della proteina p53, che normalmente determina un arresto del ciclo cellulare in G1, può portare una sopravvivenza inappropriata di cellule danneggiate e geneticamente instabili ed essere causa dell’insorgenza di una varietà di tumori.
14.5.1. Marcatori tumorali
Un marcatore (o marker) tumorale è, in senso lato, una qualsiasi sostanza correlabile alla presenza o all’evoluzione di una neoplasia. Un marcatore tumorale può (1) essere prodotto esclusivamente dalle cellule neoplastiche, (2) essere presente sia nel tessuto sano che nel tumore, ma essere espresso da quest’ultimo in maggiori quantità, o (3) essere conseguente all’interazione della neoplasia con il tessuto ospite. Quest’ultimo gruppo di marcatori comprende alcuni indici ematochimici (velocità di eritrosedimentazione, proteine della fase acuta, enzimi epatici ed ossei, etc.), la cui variazione è generalmente di limitato interesse clinico, in quanto evidente solo in uno stadio avanzato della malattia. La definizione di marcatore tumorale resta perciò convenzionalmente limitata, il più delle volte, alle sole sostanze presenti nel tumore o rilasciate da esso, che sono più specificamente legate al suo sviluppo e rappresentano un segnale precoce della presenza della neoplasia.
Con il termine “marcatore genetico” viene indicato un marcatore tumorale che
riguarda un riarrangiamento intracromosomico (per mutazione, delezione,
inversione o amplificazione) o intercromosomico (per traslocazione) della
struttura del DNA della cellula neoplastica. Con il termine “marcatore
circolante (o di secrezione)”
![]() viene indicata una sostanza secreta dal tumore, che è in grado di raggiungere
concentrazioni misurabili nei liquidi corporei prelevati da una sede di solito
lontana dalla localizzazione del tumore stesso. Un “marcatore di superficie” è
infine una sostanza espressa sulla superficie del tumore e perciò evidenziabile
sul campione bioptico mediante tecniche istochimiche. Lo studio dei marcatori di
superficie è di pertinenza dell’istopatologo ed esula perciò dalla presente
trattazione.
viene indicata una sostanza secreta dal tumore, che è in grado di raggiungere
concentrazioni misurabili nei liquidi corporei prelevati da una sede di solito
lontana dalla localizzazione del tumore stesso. Un “marcatore di superficie” è
infine una sostanza espressa sulla superficie del tumore e perciò evidenziabile
sul campione bioptico mediante tecniche istochimiche. Lo studio dei marcatori di
superficie è di pertinenza dell’istopatologo ed esula perciò dalla presente
trattazione.
14.5.1a. Marcatori genetici
L’utilità clinica dei marcatori genetici risiede (1) nella tipizzazione diagnostica delle malattie neoplastiche, (2) nel monitoraggio del clone neoplastico durante il trattamento antitumorale e (3) nella identificazione di categorie prognostiche e di gruppi omogenei per risposta a trattamenti specifici.
Lo studio dei marcatori genetici riveste una particolare importanza in campo ematologico (vedi Par. 7.1.4b). L’analisi dei riarrangiamenti dei geni delle immunoglobuline e dei recettori delle cellule T (TCR, T cell Receptors) costituisce una parte importante nella diagnosi routinaria delle affezioni linfoproliferative, consentendo di identificare l’appartenenza B- o T-linfoide di una popolazione cellulare, di testimoniare la sua origine clonale e di ottenere un marcatore specifico. Tra gli esempi più significativi di traslocazioni coinvolte nella patogenesi delle leucemie acute mieloblastiche vi sono quelle che riguardono il gene HRX sul cromosoma 11 con partner localizzati su diversi cromosomi (AF4, AF9, AF17, ENL) e produzione di trascritti chimerici contenenti caratteristiche strutturali comuni ai fattori trascrizionali. Un altro esempio coinvolgente fattori trascrizionali è fornito dalla traslocazione 6:21 presente in leucemie acute mieloblastiche che derivano da sindromi mielodisplastiche. Un sottogruppo di leucemie acute mieloidi classificate per il 90% dei casi come M2 e nel 10% dei casi come M4 è invece caratterizzato dalla presenza della traslocazione 8:21. La leucemia acuta promielocitica è infine caratterizzata dalla traslocazione 15:17 che porta alla formazione del trascritto di fusione PML/RARa e ad una alterazione della via di segnalazione endogena dell’acido retinoico.
Numerosi marcatori genetici sono in relazione con tumori di interesse non ematologico. Ricordiamo, a tale proposito, gli oncogèni della famiglia myc (per l’adenocarcinoma gastrico, il carcinoma polmonare a piccole cellule, il neuroblastoma e il retinoblastoma) e ras (per l’adenocarcinoma polmonare, il cancro del colon, del pancreas e della vescica) e gli oncosoppressori BRCA1 e BRCA2 (per il cancro della mammella e dell’ovaio) e RB1 (per il retinoblastoma).
14.5.1b. Marcatori circolanti
I marcatori tumorali circolanti possono rivestire un’importanza clinica nello screening diagnostico delle neoplasie, nella valutazione prognostica antecedente l’intervento terapeutico, nell’analisi della risposta al trattamento primario e nello studio di follow-up.
La scarsa sensibilità e specificità della maggior parte dei marcatori tumorali porta a livelli inaccettabili il rapporto costi/benefici nelle indagini di screening su larga scala. Le indagini di screening mediante l’uso di marcatori circolanti specifici sono pertanto limitate a sottopopolazioni dove il rischio di insorgenza del tumore è particolarmente elevato, mentre, nella maggior parte dei casi, la diagnosi di una neoplasia primitiva in un paziente asintomatico si basa quasi esclusivamente sulla valutazione clinica e sul risultato degli esami radiologici e bioptici. A volte, le concentrazioni sieriche del marcatore circolante si correlano bene con la massa tumorale e più elevate sono al momento della diagnosi peggiore è la prognosi. La riduzione della concentrazione di un marcatore circolante può fornire una indicazione sull’efficacia della terapia, sull’estensione della malattia residua e quindi sulla necessità di un ulteriore intervento; tuttavia la riduzione di una marcatore tumorale a livelli indosabili non implica necessariamente la guarigione dalla malattia e può, in taluni casi, anche coesistere con la presenza di un tumore di relativamente grosse dimensioni, rivelabile radiologicamente.
L’antigene carcinoembrionale (CEA) è una glicoproteina di circa 200 kDa coinvolta nei meccanismi di adesione cellulare. E’ presente nel siero a concentrazioni inferiori a 5 ng/mL ed a concentrazioni più elevate in numerose malattie neoplastiche e non-neoplastiche (epatopatie benigne, malattie infiammatorie gastrointestinali, malattie infettive, lesioni traumatiche, collagenopatie, nefropatie, tabagismo). Elevati livelli sierici di CEA sono dimostrabili nel 5% dei pazienti con carcinoma del colon-retto in stadio A, nel 25% dei pazienti in stadio B, nel 45% dei pazienti in stadio C e nel 65% dei pazienti con metastasi a distanza. A causa della bassa incidenza di valori elevati di CEA negli stadi precoci della malattia tumorale e della scarsa specificità dell’analisi, il dosaggio di questo marcatore non è utile nello screening della popolazione generale né della popolazione ad alto rischio per tumori del colon-retto, come i soggetti con poliposi familiare del colon o con rettocolite ulcerosa. L’utilità del dosaggio del CEA risiede quindi solo nella valutazione prognostica e nel monitoraggio dei tumori del colon-retto in uno stadio avanzato di sviluppo (elevati valori sierici di CEA a 4-5 settimane dalla completa resezione del carcinoma rappresentano un importante fattore prognostico sfavorevole, mentre la riduzione dei livelli di CEA dopo chemioterapia o radioterapia sono associati ad una maggiore sopravvivenza dei pazienti).
L’antigene prostatico specifico (PSA) è una proteasi glicoproteica di circa 34 kDa che deriva dal tessuto epiteliale prostatico differenziato ed è escreta in condizioni normali nel liquido seminale, che contribuisce a mantenere fluido. Il PSA è presente nel siero della maggior parte dei soggetti di sesso maschile e tende ad aumentare con l’età e nell’ipertrofia prostatica benigna. Usando un valore di cut-off di 10 ng/mL, circa il 30% dei pazienti affetti da carcinoma prostatico al primo stadio e più dell’80% di quelli con tumore metastatizzato hanno valori elevati di PSA. Convenzionalmente, per scoprire una neoplasia ancora confinata alla prostata è richiesto il superamento di un valore soglia di 4 ng/mL. Diverse strategie sono state adottate per ridurre l’incidenza di falsi positivi nelle indagini di screening, fra le quali la misurazione del PSA density (rapporto tra il livello sierico di PSA e il volume prostatico valutato mediante ecografia transrettale) e del PSA libero (non legato all’a1-antichimotripsina, vedi Par. 8.3.2), in quanto i carcinomi prostatici si associano generalmente ad una riduzione della quota libera di questo marcatore tumorale. La determinazione del PSA è risultata inoltre utile per evidenziare residui del tumore dopo trattamento chirurgico o radioterapico o per valutare la risposta all’ormonoterapia e alla chemioterapia.
La gonadotropina corionica (hCG,
vedi Par. 10.7.1) e, in
particolare, la sua subunità β
rivestono un ruolo determinante nella diagnosi e nel trattamento dei tumori
germinali del testicolo e del coriocarcinoma gestazionale
![]() . Per quanto
riguarda i tumori del testicolo, la gonadotropina corionica è presente in tutti
i pazienti con coriocarcinoma e nel 40-60% dei pazienti con carcinoma
embrionale. Il rischio di comparsa di un coriocarcinoma gestazionale è molto
basso (0,003%) dopo una normale gravidanza a termine, ma diviene pari a circa il
3% dopo una gravidanza molare, che ha di per sé una prevalenza di 1:2000
gravidanze in Europa; per tale motivo la determinazione di questo marcatore
tumorale si è dimostrata utile nello screening delle donne affette da
questa patologia. Poiché il tumore origina dal trofoblasto placentare, di solito
produce hCG in quantità proporzionale alla propria massa; il dosaggio della
gonadotropina corionica nel plasma e nelle urine è pertanto uno dei fattori
presi in considerazione nella valutazione prognostica della paziente.
. Per quanto
riguarda i tumori del testicolo, la gonadotropina corionica è presente in tutti
i pazienti con coriocarcinoma e nel 40-60% dei pazienti con carcinoma
embrionale. Il rischio di comparsa di un coriocarcinoma gestazionale è molto
basso (0,003%) dopo una normale gravidanza a termine, ma diviene pari a circa il
3% dopo una gravidanza molare, che ha di per sé una prevalenza di 1:2000
gravidanze in Europa; per tale motivo la determinazione di questo marcatore
tumorale si è dimostrata utile nello screening delle donne affette da
questa patologia. Poiché il tumore origina dal trofoblasto placentare, di solito
produce hCG in quantità proporzionale alla propria massa; il dosaggio della
gonadotropina corionica nel plasma e nelle urine è pertanto uno dei fattori
presi in considerazione nella valutazione prognostica della paziente.
La α-fetoproteina
(AFP)
![]() , oltre ad
essere prodotta durante la gravidanza, è presente in piccole quantità nel siero
normale (< 10 ng/mL). Livelli elevati in età adulta (> 20 ng/mL) sono associati
a malattie neoplastiche (carcinoma epatocellulare, gastrico, pancreatico o
polmonare, tumori delle cellule germinali del testicolo, ovaio e delle strutture
della linea mediana, inclusi il mediastino e la ghiandola pineale) o
non-neoplastiche (epatite virale, epatite cronica, cirrosi). Lo screening
del carcinoma epatocellulare attraverso la determinazione della AFP può essere
giustificato nei gruppi ad alto rischio, come i portatori di virus dell’epatite
B e C nei territori dove il carcinoma epatocellulare è più diffuso, poiché la
diagnosi precoce può consentire la completa escissione del tumore. Il 60-70% dei
pazienti con tumori germinali non seminomatosi
, oltre ad
essere prodotta durante la gravidanza, è presente in piccole quantità nel siero
normale (< 10 ng/mL). Livelli elevati in età adulta (> 20 ng/mL) sono associati
a malattie neoplastiche (carcinoma epatocellulare, gastrico, pancreatico o
polmonare, tumori delle cellule germinali del testicolo, ovaio e delle strutture
della linea mediana, inclusi il mediastino e la ghiandola pineale) o
non-neoplastiche (epatite virale, epatite cronica, cirrosi). Lo screening
del carcinoma epatocellulare attraverso la determinazione della AFP può essere
giustificato nei gruppi ad alto rischio, come i portatori di virus dell’epatite
B e C nei territori dove il carcinoma epatocellulare è più diffuso, poiché la
diagnosi precoce può consentire la completa escissione del tumore. Il 60-70% dei
pazienti con tumori germinali non seminomatosi
![]() ha livelli
elevati di α-fetoproteina e il 50-60%
livelli elevati della subunità β della
gonadotropina corionica e, se misurati simultaneamente, questi due marcatori
tumorali sono positivi in circa il 90% dei casi. In seguito all’orchiectomia, il
persistente aumento di AFP e β-HCG
suggerisce la presenza di un tumore residuo, ma la normalizzazione dei
marcatori, che erano precedentemente elevati, non sempre indica l’eradicazione
della neoplasia. D’altra parte livelli elevati di
a-fetoproteina possono persistere per
molti mesi dopo un trattamento chemioterapico senza necessariamente indicare la
presenza di una neoplasia residua attiva.
ha livelli
elevati di α-fetoproteina e il 50-60%
livelli elevati della subunità β della
gonadotropina corionica e, se misurati simultaneamente, questi due marcatori
tumorali sono positivi in circa il 90% dei casi. In seguito all’orchiectomia, il
persistente aumento di AFP e β-HCG
suggerisce la presenza di un tumore residuo, ma la normalizzazione dei
marcatori, che erano precedentemente elevati, non sempre indica l’eradicazione
della neoplasia. D’altra parte livelli elevati di
a-fetoproteina possono persistere per
molti mesi dopo un trattamento chemioterapico senza necessariamente indicare la
presenza di una neoplasia residua attiva.
Il CA125 è una glicoproteina che risulta elevata nel siero di pazienti con tumori ovarici non germinali (in particolare nel cistoadenocarcinoma), adenocarcinomi non ginecologici e molte altre affezioni benigne. Questo marcatore non possiede la specificità diagnostica necessaria per poter essere utilizzato a scopo di screening. In associazione all’esame clinico e all’ultrasonografia, le concentrazioni sieriche di Ca125 sono utili a stabilire se la terapia dei tumori ovarici è appropriata.
Il CA19-9 è un antigene che reagisce con un anticorpo monoclonale specifico ottenuto nei topi sensibilizzati contro una linea cellulare umana derivata da cellule di carcinoma colon-rettale. Le concentrazioni sieriche di Ca19-9 risultano elevate in più dell’80% dei pazienti affetti da adenocarcinoma del pancreas esocrino, in quasi il 50% dei pazienti con carcinoma dello stomaco e nel 20-30% dei pazienti con cancro del colon-retto, ma solo in una piccola percentuale dei soggetti con pancreopatia cronica benigna. Purtroppo questo marcatore ha scarso valore clinico in quanto aumenta solo in una fase tardiva della malattia neoplastica, quando vi sono scarse possibilità terapeutiche.
Altri marcatori circolanti, che possono essere utilizzati nella pratica clinica, sono la calcitonina (un marcatore altamente specifico per il raro carcinoma midollare della tiroide), la tireoglobulina (utile per evidenziare la presenza di tessuto tiroideo residuo o di recidive in pazienti tiroidectomizzati) e l’enolasi neurono-specifica (di limitata utilità come indice prognostico in caso di microcitoma).
14.6. TEST DIAGNOSTICI
In base alle attuali raccomandazioni, i test genetici dovrebbero: (1) essere eseguiti sempre sotto indicazione di un consultorio genetico in modo da informare adeguatamente il paziente sulle implicazioni derivanti dai risultati, (1) essere condotti su base volontaria senza alcuna pressione sociale o incentivo finanziario, dando luogo a risultati riservati in modo da evitare abusi, (3) essere limitati a quelle patologie per le quali esista la possibilità di un intervento positivo in forma di terapia o di programmazione familiare.
14.6.1. Screening di massa
Tutti i programmi di screening si trovano a
fronteggiare complessi problemi di carattere etico ed
organizzativo e possono essere responsabili, se strutturati male,
di notevoli danni psicologici e sociali per i pazienti
![]() .
Inoltre, per avere successo un programma di screening di
massa deve riguardare errori genetici sufficientemente diffusi
nella comunità.
.
Inoltre, per avere successo un programma di screening di
massa deve riguardare errori genetici sufficientemente diffusi
nella comunità.
La frequenza relativa delle malattie genetiche varia
marcatamente nelle diverse popolazioni. Per esempio, la fibrosi cistica
![]() e il deficit di
α1-antitripsina
(vedi Par. 8.3.1)
sono più frequenti nell’Europa settentrionale, le patologie eritrocitarie (talassemia, anemia
falciforme, deficit di glucosio 6-fosfato deidrogenasi; vedi
Par. 7.1.3a) sono più
comuni nelle popolazioni mediterranee ed africane, la malattia di
Tay-Sachs
e il deficit di
α1-antitripsina
(vedi Par. 8.3.1)
sono più frequenti nell’Europa settentrionale, le patologie eritrocitarie (talassemia, anemia
falciforme, deficit di glucosio 6-fosfato deidrogenasi; vedi
Par. 7.1.3a) sono più
comuni nelle popolazioni mediterranee ed africane, la malattia di
Tay-Sachs
![]() è presente soprattutto fra gli ebrei Ashkenazi.
L’alta prevalenza di una malattia ereditaria in popolazioni
relativamente ristrette può essere dovuta al loro isolamento per
ragioni geografiche, razziali o culturali, come è avvenuto nel
Sudafrica per la porfiria variegata
è presente soprattutto fra gli ebrei Ashkenazi.
L’alta prevalenza di una malattia ereditaria in popolazioni
relativamente ristrette può essere dovuta al loro isolamento per
ragioni geografiche, razziali o culturali, come è avvenuto nel
Sudafrica per la porfiria variegata
![]() . Le malattie
autosomiche recessive, che risultano relativamente diffuse in
ampie popolazione, potrebbero invece essere espressione di un
polimorfismo bilanciato, in cui lo svantaggio negli omozigoti è
bilanciato dai vantaggi acquisiti da un gran numero di
eterozigoti. Nel caso dell’anemia falciforme, il vantaggio selettivo è
dovuto ad una maggiore resistenza alla malaria da Plasmodium
falciparum. Nel caso dell’emocromatosi ereditaria
. Le malattie
autosomiche recessive, che risultano relativamente diffuse in
ampie popolazione, potrebbero invece essere espressione di un
polimorfismo bilanciato, in cui lo svantaggio negli omozigoti è
bilanciato dai vantaggi acquisiti da un gran numero di
eterozigoti. Nel caso dell’anemia falciforme, il vantaggio selettivo è
dovuto ad una maggiore resistenza alla malaria da Plasmodium
falciparum. Nel caso dell’emocromatosi ereditaria
![]() , uno dei più comuni
geni legati allo sviluppo di una malattia presente nelle
popolazioni di origine europea, il vantaggio selettivo sarebbe
stato connesso ad una relativa protezione nei riguardi di un deficit
di ferro. L’elevata prevalenza di un allele mutante per l’isoenzima
muscolare dell’adenilato deaminasi, causa di una lieve
miopatia metabolica, è in relazione con una migliore prognosi in
caso di insufficienza cardiaca congestizia. Soggetti con una
mutazione spontanea del
recettore CCR5 per le chemochine sono
resistenti all’infezione da HIV in quanto questa proteina
rappresenta un corecettore di fusione per l’infezione virale.
Questi esempi mostrano quanto sia difficile formulare un giudizio
a priori nei riguardi di una mutazione del genoma umano e pongono
seri dubbi sulla effettiva utilità dei programmi di eugenetica.
, uno dei più comuni
geni legati allo sviluppo di una malattia presente nelle
popolazioni di origine europea, il vantaggio selettivo sarebbe
stato connesso ad una relativa protezione nei riguardi di un deficit
di ferro. L’elevata prevalenza di un allele mutante per l’isoenzima
muscolare dell’adenilato deaminasi, causa di una lieve
miopatia metabolica, è in relazione con una migliore prognosi in
caso di insufficienza cardiaca congestizia. Soggetti con una
mutazione spontanea del
recettore CCR5 per le chemochine sono
resistenti all’infezione da HIV in quanto questa proteina
rappresenta un corecettore di fusione per l’infezione virale.
Questi esempi mostrano quanto sia difficile formulare un giudizio
a priori nei riguardi di una mutazione del genoma umano e pongono
seri dubbi sulla effettiva utilità dei programmi di eugenetica.
Lo screening dei portatori (eterozigoti) di difetti
ereditari recessivi è, allo stato attuale, generalmente limitato
all’identificazione di emoglobinopatie e di poche altre
condizioni a rischio in minoranze etniche con alta incidenza
della patologia (vedi Par. 14.6.2). Lo screening
neonatale per l’identificazione degli individui affetti da
fenilchetonuria ![]() e ipotiroidismo congenito (vedi
Par. 10.4) viene attualmente
eseguito usando test biochimici convenzionali su gocce di
sangue essiccato ottenuto da punture sul tallone nelle prime due
settimane di vita (vedi Par. 1.5.1a). Questi stessi campioni possono essere anche
utilizzati per eseguire ulteriori analisi biochimiche e molecolari al
fine di identificare altre malattie ereditarie, quando questa
indagine risulti vantaggiosa per la disponibilità di un
trattamento efficace
e ipotiroidismo congenito (vedi
Par. 10.4) viene attualmente
eseguito usando test biochimici convenzionali su gocce di
sangue essiccato ottenuto da punture sul tallone nelle prime due
settimane di vita (vedi Par. 1.5.1a). Questi stessi campioni possono essere anche
utilizzati per eseguire ulteriori analisi biochimiche e molecolari al
fine di identificare altre malattie ereditarie, quando questa
indagine risulti vantaggiosa per la disponibilità di un
trattamento efficace ![]() .
.
14.6.2. Screening prenatale
Uno screening prenatale per le malattie ereditarie è
indicato in presenza di fattori di rischio generici, specifici o
etnici. Sono fattori generici di rischio l’età materna
superiore a 35 anni e la positività al Tri-test. Sono
fattori di rischio specifici i precedenti aborti o morti
neonatali, i precedenti figli con aberrazioni cromosomiche, una
patologia cromosomica nei collaterali, una traslocazione
bilanciata in uno dei genitori, una sindrome da geni contigui (vedi
Par. 14.1.4) o da imprinting genomico
(vedi Par. 14.1.3), una malattia
ereditaria da
difetto di riparo del DNA, una malattia genetica
del metabolismo o da mutazioni dinamiche (vedi Par. 14.2.3), l’esposizione ad agenti teratogeni (radiazioni,
litio, anticonvulsivanti
![]() ) o una infezione da rosolia, toxoplasmosi o citomegalovirus. Sono fattori di rischio etnico
quelle malattie ad alta prevalenza in alcune popolazioni, come ad esempio la talassemia nelle popolazioni mediterranee,
cinesi e dell’Asia del sud, l’anemia falciforme nelle popolazioni
mediteranee, negro-africane, arabe, indiane e pakistane (vedi
Par.7.2.1), la malattia di Tay-Sachs
) o una infezione da rosolia, toxoplasmosi o citomegalovirus. Sono fattori di rischio etnico
quelle malattie ad alta prevalenza in alcune popolazioni, come ad esempio la talassemia nelle popolazioni mediterranee,
cinesi e dell’Asia del sud, l’anemia falciforme nelle popolazioni
mediteranee, negro-africane, arabe, indiane e pakistane (vedi
Par.7.2.1), la malattia di Tay-Sachs
![]() fra gli ebrei Ashkenazi e
la tirosinosi fra i canadesi del Quebec
fra gli ebrei Ashkenazi e
la tirosinosi fra i canadesi del Quebec
![]() .
.
Il dosaggio simultaneo della concentrazione dell’α-fetoproteina, della gonadotropina
corionica e dell’estriolo non coniugato nel plasma materno
intorno alla sedicesima settimana di gestazione costituisce il Tri-test
(o test triplo) che, in combinazione con l’età
ed il peso della madre, esprime in termini di punteggio il
rischio che il feto sia affetto da sindrome di Down
![]() .
Il rischio aumenta con la diminuzione della concentrazione dell’a-fetoproteina e dell’estriolo non
coniugato e con l’aumento della gonadotropina corionica (vedi
Par. 10.7.4). Il Tri-test
non è tuttavia in alcun caso diagnostico della condizione
morbosa, ma permette solo una migliore selezione delle donne a
cui proporre l’amniocentesi. La convinzione che il rischio
di una sindrome di Down possa dipendere esclusivamente dall’età
materna o da fattori di rischio specifici ha infatti portato all’esecuzione
di indagini invasive per la raccolta di campioni in tutte le
donne di età superiore a 35 anni. Purtroppo, questo approccio
non ha portato ad una riduzione significativa del numero dei nati
affetti dalla sindrome, nonostante il 3% delle donne gravide sia
stato sottoposto all’indagine bioptica. Il fenomeno è
probabilmente dovuto al fatto che, sebbene il rischio aumenti con
l’età materna, il 70% dei bambini con sindrome di Down
nasce da donne di età inferiore ai 35 anni, meno propense a
sottoporsi all’esame per timore di un aborto.
.
Il rischio aumenta con la diminuzione della concentrazione dell’a-fetoproteina e dell’estriolo non
coniugato e con l’aumento della gonadotropina corionica (vedi
Par. 10.7.4). Il Tri-test
non è tuttavia in alcun caso diagnostico della condizione
morbosa, ma permette solo una migliore selezione delle donne a
cui proporre l’amniocentesi. La convinzione che il rischio
di una sindrome di Down possa dipendere esclusivamente dall’età
materna o da fattori di rischio specifici ha infatti portato all’esecuzione
di indagini invasive per la raccolta di campioni in tutte le
donne di età superiore a 35 anni. Purtroppo, questo approccio
non ha portato ad una riduzione significativa del numero dei nati
affetti dalla sindrome, nonostante il 3% delle donne gravide sia
stato sottoposto all’indagine bioptica. Il fenomeno è
probabilmente dovuto al fatto che, sebbene il rischio aumenti con
l’età materna, il 70% dei bambini con sindrome di Down
nasce da donne di età inferiore ai 35 anni, meno propense a
sottoporsi all’esame per timore di un aborto.
Lo screening prenatale è indicato anche per l’individuazione
di un limitato numero di malattie genetiche nelle quali l’efficacia
del trattamento è funzione della precocità della diagnosi e
dell’inizio della terapia. Bisogna ricordare a questo
proposito la possibilità di somministrare desametasone per via
orale alla madre a partire della settima settimana di gestazione
in caso di deficit di 21-idrossilasi (il trattamento viene
sospeso se il feto risulta di sesso maschile e continuato se di
sesso femminile; vedi Par. 10.5.1d), di eseguire un trapianto in utero di cellule staminali
ematopoietiche paterne nei feti con immunodeficienza combinata grave X-recessiva
(vedi Par. 8.6.2a), di prescrivere
al neonato con deficit di galattosio-1-fosfato uridiltransferasi
![]() una dieta
povera di galattosio immediatamente dopo la nascita
una dieta
povera di galattosio immediatamente dopo la nascita
![]() .
.
Esiste una notevole diatriba sui rapporti tra la medicina ed i test genetici prenatali. L’accettabilità o meno dei test genetici quando l’intenzione è di evitare la nascita di un figlio malato dipende da tanti fattori, comprese la gravità della malattia e la disponibilità di cure idonee. Poiché quest’ultimo aspetto è verosimilmente suscettibile di miglioramento col tempo, è necessario un costante aggiornamento nel campo.
14.6.3. Profilo genetico
Lo studio dei profili genetici (DNA Profiling, Genetic Fingerprinting) è particolarmente utile in medicina forense per l’identificazione di resti o tracce biologiche umane oppure per l’accertamento della paternità o della maternità.
La metodica più
frequentemente usata si basa sull’analisi
dei polimorfismi di lunghezza in microsatelliti contenenti ripetizioni (più
o meno ampie nei diversi alleli) di brevi sequenze di 3-5 basi (STR,
Short Tandem
Repeats). I
loci STR sono localizzati mediante l’uso
di primer
specifici ed amplificati mediante PCR (vedi Par. 14.6.6b);
i frammenti di DNA sono quindi separati ed identificati mediante
elettroforesi capillare o su gel. Poiché ogni singolo locus STR presenta un
numero limitato di alleli (generalmente ogni allele è riscontrabile nel
5-20% della popolazione), sono esaminati di norma 13-20 loci STR
simultaneamente in modo da aumentare enormemente il potere discriminante
della metodica ![]() .
.
Recentemente è stata creata una serie di primer per analizzare i polimorfismi sul cromosoma Y (Y-STR), per permettere di identificare il profilo genetico maschile in tracce biologiche contenenti DNA di entrambi i sessi. Nel caso in cui il materiale in esame sia fortemente degradato, può essere invece conveniente ricostruire il profilo genetico utilizzando il DNA mitocondriale, che è presente in copie multiple ed è generalmente meglio conservato; a tale scopo si può adoperare del materiale proveniente dai capelli, dalle ossa o dai denti.
14.6.4. Metodi di determinazione
La diagnosi iniziale di una malattia genetica si basa di solito sull’anamnesi, sulle manifestazioni cliniche e su semplici indagini di laboratorio. La conferma della diagnosi può essere poi ottenuta mediante uno specifico test enzimatico o uno studio della proteina anomala.
Le indagini sul DNA si sono rivelate utili in un limitato
numero di casi quando (1) la proteina difettosa non è
stata ancora identificata (come nella corea di Huntington
![]() ) o è di difficile
determinazione (come nella fibrosi cistica
) o è di difficile
determinazione (come nella fibrosi cistica
![]() ), o (2) vi sono
frequenti delezioni geniche parziali o complete relativamente
semplici da identificare (come nella distrofia muscolare
), o (2) vi sono
frequenti delezioni geniche parziali o complete relativamente
semplici da identificare (come nella distrofia muscolare
![]() ).
).
Un altro vantaggio delle indagini sul DNA nella diagnosi di una malattia ereditaria è che i geni mutati sono presenti in tutte le cellule nucleate (compresi i linfociti del sangue periferico, le cellule di sfaldamento della mucosa orale, le cellule presenti nel liquido amniotico, etc.). Ciò si rivela particolarmente utile quando l’espressione genica è limitata ad organi come il fegato o il rene in quanto si può evitare di sottoporre il paziente ad un prelievo bioptico. Il principale limite delle indagini sul DNA sta nella eterogeneità delle mutazioni responsabili della malattia ereditaria perché ciò ostacola la preparazione di un test diagnostico molecolare adatto a tutte le situazioni.
14.6.4a. Diagnosi biochimica
Gran parte dei metaboliti intermedi, che risultano alterati nelle malattie genetiche, sono dosati utilizzando le metodiche già descritte nei capitoli precedenti. Le alterazioni delle attività enzimatiche associate ai difetti metabolici vengono evidenziate mediante tecniche colorimetriche o cromatografiche su fluidi biologici od estratti cellulari. Specifiche procedure sono state messe a punto per lo screening di malattie metaboliche ereditarie di più frequente riscontro.
La presenza nelle urine di un carboidrato diverso dal glucosio può essere sospettata quando il campione è positivo al test di Benedict, ma negativo ai test enzimatici per la determinazione di questo zucchero (vedi Par. 2.1.1). Il carboidrato è identificato mediante cromatografia su carta o su strato sottile e colorato con ftalato di anilina o resorcinolo.
Gli aminoacidi presenti nel siero possono essere
identificati, dopo precipitazione della componente proteica,
mediante elettroforesi o cromatografia per ripartizione (su
carta, cellulosa, gel di silice, etc.) o per scambio ionico. L’analita
viene dosato colorimetricamente in presenza di ninidrina, 1-fluoro-2,4-dinitrobenzene
o β-naftochinone sulfonato. Specifiche
procedure analitiche sono disponibili per identificare alcuni
aminoacidi di maggiore importanza clinica: la fenilalanina è
dosabile in presenza di ninidrina e sali di rame, la tirosina in
presenza di 1-nitroso-2-naftolo e acido nitrico, la cistina dopo
riduzione a cisteina in presenza di nitroprussiato di sodio o del
reattivo di Ellman (acido 5-5’-ditiobis-2-nitrobenzoico), l’istidina
con il reattivo di Pauly (acido sulfanilico), l’idrossiprolina
e la citrullina con il reattivo di Ehrlich (p-dimetilaminobenzaldeide).
E’ disponibile uno specifico e sensibile metodo enzimatico
per il dosaggio della fenilalanina e della tirosina: in presenza
della fenilalanina-ammoniaca liasi estratta da lievito, la
fenilalanina è convertita in acido trans-cinnamico (che assorbe
a 290 nm) e la tirosina in acido p-coumarico (che
assorbe a 315 nm). Sono infine disponibili alcuni test
microbiologici per gli screening su larga scala. Essi si
basano sulla capacità che l’aminoacido, presente in
eccesso nel campione, ha di permettere la crescita di batteri su
terreni in cui è presente un inibitore specifico: il test di
Guthrie (specifico per la fenilchetonuria
![]() ) utilizza un
antagonista della fenilalanina, la β-2-tienilalanina,
che inibisce la crescita di un particolare ceppo (ATCC 6051) di Bacillus
subtilis; procedure analoghe sono state messe a punto per
gli screening riguardanti la tirosinemia
) utilizza un
antagonista della fenilalanina, la β-2-tienilalanina,
che inibisce la crescita di un particolare ceppo (ATCC 6051) di Bacillus
subtilis; procedure analoghe sono state messe a punto per
gli screening riguardanti la tirosinemia
![]() , l’istidinemia,
la valinemia, l’omocistimuria
, l’istidinemia,
la valinemia, l’omocistimuria
![]() (evidenziando l’aumento
di metionina nel sangue) e la malattia ad urine a sciroppo d’acero
(evidenziando l’aumento
di metionina nel sangue) e la malattia ad urine a sciroppo d’acero
![]() (evidenziando l’aumento di leucina nel sangue).
(evidenziando l’aumento di leucina nel sangue).
14.6.4b. Diagnosi diretta della mutazione genica
La perdita di ampi settori del DNA, comprendenti centinaia di migliaia o milioni di coppie di basi con delezione di molti geni, può essere studiata mediante l’analisi dei cromosomi al microscopio ottico. Microdelezioni e duplicazioni, che interessano più geni in tratti limitati del cromosoma, possono essere evidenziate mediante ibridazione in sito con sonde fluorescenti (vedi Par. 14.6.6d). Queste metodiche sono ad esempio utilizzate nello studio delle sindromi da geni contigui (vedi Par. 14.1.4)
Delezioni più limitate, comprese all’interno di un gene
(come nei casi di distrofia muscolare di Duchenne o di Becker
![]() ),
sono identificabili mediante tecniche di amplificazione
attraverso la reazione polimerasica a catena (Par. 14.6.6b) e di ibridazione su filtro (Par.
14.6.6e). Le stesse tecniche sono utilizzabili in caso di
inserzioni, duplicazione o espansione del gene o di mutazioni
puntiformi.
),
sono identificabili mediante tecniche di amplificazione
attraverso la reazione polimerasica a catena (Par. 14.6.6b) e di ibridazione su filtro (Par.
14.6.6e). Le stesse tecniche sono utilizzabili in caso di
inserzioni, duplicazione o espansione del gene o di mutazioni
puntiformi.
Nel caso in cui il sito specifico di mutazione sia sconosciuto, è possibile usare dei metodi di screening di mutazione utilizzando eteroduplicati formati da filamenti di DNA mutante e DNA standard ibridizzati fra loro. La tecnica di discordanza di scissione permette di scindere le catene per via chimica o enzimatica a livello della sede del paio di basi discordanti e di separare ed identificare i frammenti di diversa dimensione mediante elettroforesi. Le tecniche di elettroforesi su gel a gradiente denaturante o su gel a gradiente di temperatura sfruttano il fatto che l’eteroduplicato inizia a dissociarsi ed a ridurre la sua mobilità elettroforetica precocemente, in un punto caratteristico del gradiente. Un altro metodo di screening (Single-Stranded Conformational Polymorphism; SSCP) si basa sul fatto che le sequenze di DNA a catena singola si piegano in conformazioni specifiche in modo che le sequenze di DNA standard e mutante possono venire separate mediante elettroforesi su gel di poliacrilamide. La caratterizzazione delle nuove mutazioni richiede il sequenziamento del DNA interessato (vedi Par. 14.6.6f).
14.6.4c. Analisi di linkage
E’ possibile giungere ad una diagnosi molecolare di
malattia genetica anche quando non è possibile identificare la
mutazione vera e propria, a patto che il gene sia noto o, almeno,
sia stata effettuata la sua localizzazione lungo il genoma umano
mediante mappaggio genico. Questa analisi si effettua studiando
sequenze di DNA polimorfiche
![]() che non sono di per sé causa di
malattia, ma che hanno la caratteristica di essere strettamente
associate al gene in esame o, addirittura, di essere situate al
suo interno. In questo modo le sequenze polimorfiche funzionano
come marcatori dei diversi membri di una famiglia e servono per
individuare i soggetti probabili portatori del gene alterato.
che non sono di per sé causa di
malattia, ma che hanno la caratteristica di essere strettamente
associate al gene in esame o, addirittura, di essere situate al
suo interno. In questo modo le sequenze polimorfiche funzionano
come marcatori dei diversi membri di una famiglia e servono per
individuare i soggetti probabili portatori del gene alterato.
La tecnica richiede una analisi estesa a tutti i membri disponibili della famiglia in esame allo scopo di individuare marcatori del DNA informativi, in grado cioè di identificare il gene alterato. Tuttavia, il metodo può essere inficiato (1) dalla presenza di una ricombinazione genetica fra marcatore e gene alterato, la cui frequenza è proporzionale alla distanza fra i due siti, (2) dalla eterogeneità genetica del difetto ereditario, quando più geni possono essere responsabili della stessa malattia in famiglie diverse, o (3) dalla insicurezza delle relazioni parentali biologiche fra i membri della famiglia stessa.
14.6.5. Preparazione del campione
Lo studio delle malattie genetiche può richiedere l’analisi di fluidi biologici (plasma, siero, fluido cerebrospinale, liquido amniotico, urine), campioni di sangue, biopsie o cellule in coltura.
L’analisi dei fluidi biologici è indicata quando si
sospetta la carenza o l’accumulo in essi di una proteina o
un metabolita intermedio. Il plasma e il siero sono anche usati
nello studio di alterazioni enzimatiche, come ad esempio nella malattia di Tay-Sachs
![]() e nel deficit di biotinidasi
e nel deficit di biotinidasi
![]() .
.
I globuli rossi costituiscono un materiale di facile
reperimento per lo studio non solo delle emoglobinopatie, ma
anche di diverse enzimopatie, come la sindrome di Lesch-Nyhan
![]() , la galattosemia
, la galattosemia
![]() e il deficit di diversi enzimi
della glicolisi. Anche i globuli bianchi possono essere
facilmente isolati dai pazienti e da volontari sani. I leucociti
possono essere adoperati per identificare diversi errori
metabolici, come la malattia ad urine a sciroppo d’acero
e il deficit di diversi enzimi
della glicolisi. Anche i globuli bianchi possono essere
facilmente isolati dai pazienti e da volontari sani. I leucociti
possono essere adoperati per identificare diversi errori
metabolici, come la malattia ad urine a sciroppo d’acero
![]() , acidosi metilmalonica
, acidosi metilmalonica
![]() , alcune
glicogenosi e
gran parte delle
malattie lisosomiali d’accumulo.
, alcune
glicogenosi e
gran parte delle
malattie lisosomiali d’accumulo.
Le biopsie del fegato, della mucosa intestinale, del muscolo e della tiroide possono essere effettuate con modesto rischio per il paziente e sono indicate per ottenere informazioni non disponibili altrimenti.
Lo studio di cellule in coltura (fibroblasti e linfoblasti trasformati con il virus di Epstein-Barr) ha il vantaggio (1) di poter seguire l’incorporazione e il metabolismo intracellulare di precursori marcati, (2) di poter ripetere ed ampliare gli studi senza dover ricorrere nuovamente al paziente, (3) di poter analizzare le mutazioni a livello del DNA e dell’RNA, (4) di poter comparare facilmente fra loro i risultati ottenuti con diversi pazienti.
14.6.5a. Diagnosi prenatale
La diagnosi genetica preimpianto può essere eseguita prelevando uno dei due
corpi polari da un uovo
![]() o un blastomero da un embrione, che è derivato da una
fecondazione in vitro ed ha raggiunto lo stadio di morula a otto cellule (terzo
giorno di sviluppo; Fig. 14.2). Nel primo caso si possono ovviamente studiare solo i difetti genetici
di derivazione materna, mentre nel secondo caso si può ottenere un quadro
complessivo delle anomalie presenti nell’embrione, che viene impiantato in utero.
Questo tipo di analisi ha lo scopo di verificare se il prodotto del concepimento
è esente o portatore sano del carattere ereditario recessivo fortemente
invalidante e dall’esito letale oggetto della ricerca (fibrosi cistica
o un blastomero da un embrione, che è derivato da una
fecondazione in vitro ed ha raggiunto lo stadio di morula a otto cellule (terzo
giorno di sviluppo; Fig. 14.2). Nel primo caso si possono ovviamente studiare solo i difetti genetici
di derivazione materna, mentre nel secondo caso si può ottenere un quadro
complessivo delle anomalie presenti nell’embrione, che viene impiantato in utero.
Questo tipo di analisi ha lo scopo di verificare se il prodotto del concepimento
è esente o portatore sano del carattere ereditario recessivo fortemente
invalidante e dall’esito letale oggetto della ricerca (fibrosi cistica
![]() , distrofia muscolare tipo Duchenne
, distrofia muscolare tipo Duchenne
![]() , malattia di Tay-Sachs
, malattia di Tay-Sachs
![]() ).
).

Fig. 14.2. Prelievo di un corpo polare da un uovo (a sinistra) e di un blastomero da una morula di otto cellule (a destra). Entrambe le biopsie sono eseguite con l’ausilio di un microscopio manipolatore ancorando le cellule con una pipetta dal lato opposto rispetto al prelievo.
La biopsia dei villi coriali viene eseguita di preferenza nel periodo compreso tra il 70° (decima settimana) e il 91° (tredicesima settimana) giorno dall’inizio dell’ultima mestruazione, quando il chorion frondosum è facilmente identificabile all’esame ecografico. La scelta di questa finestra consente di giungere ad una diagnosi durante il primo trimestre, evitando nel contempo le settimane iniziali di gravidanza che hanno una abortività spontanea molto più elevata. Quando la biopsia è eseguita per via transaddominale, il rischio di aborto è pari al 2,3%. La via transcervicale ha un rischio più elevato (3,6%), ma permette di ottenere un campione di maggiori dimensioni; è preferibile nel caso di inserzione sul fondo dell’utero ed è di elezione quando sono presenti anse intestinali lungo la traiettoria dell’ago.
L’amniocentesi viene eseguita di norma nel periodo compreso fra la quindicesima e la diciasettesima settimana con un rischio del 0,1-1%. Una amniocentesi in epoca più precoce (tra la decima e la quattordicesima settimana) è legata alla necessità di pervenire il più rapidamente possibile alla diagnosi di anomalie cromosomiche, ma comporta un maggiore rischio di aborto (5,3%).
Il prelievo di sangue fetale mediante cordocentesi comporta una abortività di circa il 2% ed è effettuato soprattutto per esami sierologici in gravidanze a rischio di infezioni fetali.
La biopsia fetale sotto controllo ultrasonico viene eseguita per determinare affezioni genetiche cutanee, come la displasia ectodermica anidrotica, l’epidermosi bollosa, i disturbi della pigmentazione (sindrome di Chédiak-Higashi, albinismo tirosinasi-negativo) e i disturbi della cheratinizzazione (feto arlecchino, collodion baby, sindrome di Sjögren-Larsson, ipercheratosi epidermolitica generalizzata).
14.6.6. Appendice: tecniche di biologia molecolare
I metodi attualmente utilizzati in biologia molecolare sfruttano la capacità inerente alla struttura del DNA a singolo filamento di riconoscere e legarsi (ibridizzarsi) specificatamente al suo filamento complementare, nonché i numerosi enzimi di restrizione di origine batterica capaci di riconoscere e scindere specifiche sequenze nucleotidiche.
14.6.6a. Enzimi di restrizione
Gli enzimi di restrizione sono endonucleasi di origine
batterica che riconoscono e scindono il DNA a doppia elica a
livello di "siti di restrizione" ben definiti. Questi
siti sono generalmente costituiti da una sequenza palindromica
![]() di 4-6 basi. Gli enzimi di restrizione
idrolizzano il legame fosfodiesterico su ambedue le eliche. Le
caratteristiche e le modalità del riconoscimento e della
scissione del DNA sono diverse nei diversi enzimi, che vengono
denominati con sigle che alludono alla specie batterica da cui
sono stati isolati. Alcuni enzimi riconoscono solo determinate
sequenze nucleotidiche, altri sono meno specifici e riconoscono
sequenze in cui una o più basi del sito di restrizione possono
essere sostituite da basi diverse. Un’altra differenza
importante riguarda la modalità di scissione del DNA: alcuni
enzimi scindono entrambe le catene del DNA in modo tale da non
lasciare basi non appaiate, altri operano tagli sfalsati che
lasciano due o quattro nucleotidi liberi in ciascuna estremità,
capaci di riappaiarsi e complementarsi tra loro (estremità
coesive). La lunghezza media dei frammenti prodotti dalla
scissione del genoma mediante un enzima di restrizione dipende
dalla frequenza con cui un particolare sito di restrizione
compare nel DNA e questa dipende, a sua volta, dalla lunghezza
della sequenza di riconoscimento.
di 4-6 basi. Gli enzimi di restrizione
idrolizzano il legame fosfodiesterico su ambedue le eliche. Le
caratteristiche e le modalità del riconoscimento e della
scissione del DNA sono diverse nei diversi enzimi, che vengono
denominati con sigle che alludono alla specie batterica da cui
sono stati isolati. Alcuni enzimi riconoscono solo determinate
sequenze nucleotidiche, altri sono meno specifici e riconoscono
sequenze in cui una o più basi del sito di restrizione possono
essere sostituite da basi diverse. Un’altra differenza
importante riguarda la modalità di scissione del DNA: alcuni
enzimi scindono entrambe le catene del DNA in modo tale da non
lasciare basi non appaiate, altri operano tagli sfalsati che
lasciano due o quattro nucleotidi liberi in ciascuna estremità,
capaci di riappaiarsi e complementarsi tra loro (estremità
coesive). La lunghezza media dei frammenti prodotti dalla
scissione del genoma mediante un enzima di restrizione dipende
dalla frequenza con cui un particolare sito di restrizione
compare nel DNA e questa dipende, a sua volta, dalla lunghezza
della sequenza di riconoscimento.
Gli enzimi di restrizione possono essere utilizzati per analizzare le diversità molecolari e identificare l’individualità delle sequenze di DNA. Mutazioni lungo il DNA possono infatti creare o distruggere siti di riconoscimento per gli enzimi di restrizione e determinare una variazione nella lunghezza dei frammenti prodotti da questi enzimi (Restriction Fragment Lenght Polimorphism; RFLP). I frammenti di DNA possono essere separati elettroforeticamente ed identificati mediante l’uso di sonde specifiche (vedi Par. 14.6.6e).
14.6.6b. Reazione polimerasica a catena (PCR)
Questo metodo è in grado di clonare in vitro il DNA
mediante l’uso di una DNA-polimerasi e di due primer
oligonucleotidici complementari a sequenze che fiancheggiano la
regione da analizzare nel DNA bersaglio, uno su ogni catena
![]() .
La sequenza bersaglio è amplificata in modo esponenziale
attraverso cicli ripetuti di copiatura enzimatica. Nel primo
ciclo, la catena a doppia elica viene denaturata a 92-96° C e
poi raffreddata a 55-60° C in modo che i primer possano
allinearsi alle loro sequenze complementari sul DNA bersaglio. A
questo punto può aver luogo l’estensione dei primer
mediante la DNA-polimerasi, usando nucleotidi aggiunti e il DNA
bersaglio come stampo. Il DNA viene nuovamente denaturato,
permettendo ai primer in eccesso di allinearsi sia alle
catene neoformate sia a quelle originali e il processo di sintesi
viene ripetuto. In questo modo, in circa 30 cicli è possibile
ottenere una amplificazione del DNA bersaglio di oltre un milione
di volte. L’uso di un enzima termostabile (Taq-polimerasi)
evita la necessità di aggiungere enzima fresco ad ogni ciclo e
rende possibile la reazione ad alta temperatura (72° C).
.
La sequenza bersaglio è amplificata in modo esponenziale
attraverso cicli ripetuti di copiatura enzimatica. Nel primo
ciclo, la catena a doppia elica viene denaturata a 92-96° C e
poi raffreddata a 55-60° C in modo che i primer possano
allinearsi alle loro sequenze complementari sul DNA bersaglio. A
questo punto può aver luogo l’estensione dei primer
mediante la DNA-polimerasi, usando nucleotidi aggiunti e il DNA
bersaglio come stampo. Il DNA viene nuovamente denaturato,
permettendo ai primer in eccesso di allinearsi sia alle
catene neoformate sia a quelle originali e il processo di sintesi
viene ripetuto. In questo modo, in circa 30 cicli è possibile
ottenere una amplificazione del DNA bersaglio di oltre un milione
di volte. L’uso di un enzima termostabile (Taq-polimerasi)
evita la necessità di aggiungere enzima fresco ad ogni ciclo e
rende possibile la reazione ad alta temperatura (72° C).
La PCR è una metodica estremamente sensibile ed è in grado di amplificare quantità molto piccole di DNA. Per questo motivo è sempre necessario prendere le opportune precauzioni per evitare contaminazioni da parte di DNA estraneo (per esempio dell’operatore). Una maggiore specificità può essere ottenuta mediante la tecnica dell’hot start che evita allineamenti aspecifici dei primer iniziando la reazione enzimatica ad una temperatura superiore a quella dell’allineamento stesso. Un’altra tecnica è quella dei primer nested in cui una prima amplificazione del DNA è seguita da una seconda amplificazione che utilizza una coppia di primer interni rispetto a quelli usati la prima volta.
La tecnica della amplificazione allele-specifica (Amplification Refractory Mutation System, ARMS) sfrutta la specificità dei primer usati per la PCR e permette di identificare mutazioni anche puntiformi del genoma. Con questa tecnica, il materiale da analizzare viene diviso in due frazioni; nella prima frazione viene utilizzato un primer capace di ibridizzare con la sequenza di DNA dove è presente la mutazione, mentre nella seconda frazione viene utilizzato un primer capace di ibridizzare con l’analoga regione del DNA dei soggetti di controllo. In tal modo, i primer complementari alla sequenza nucleotidica di controllo potranno amplificare le catene polinucleotidiche solo se è presente il DNA "normale" (wild type), mentre, se è stato utilizzato il primer corrispondente alla sequenza mutata, sarà solo quest’ultima che potrà essere amplificata. Ovviamente, nel caso di soggetti eterozigoti, l’amplificazione del DNA sarà possibile con entrambi i primer.
Per evidenziare rapidamente delezioni o duplicazioni su geni di grandi dimensioni si può adoperare una PCR modificata (Multiplex Polymerase Chain Reaction) consistente nell’uso contemporaneo di più coppie di primer in modo da produrre frammenti amplificati (amplicons) di varie dimensioni, corrispondenti a diverse regioni del DNA. Dopo l’amplificazione, i frammenti di diversa lunghezza sono separati mediante elettroforesi. Questa tecnica è risultata particolarmente utile nello studio dei profili genetici (vedi Par. 14.6.3).
14.6.6c. Sonde molecolari
Le sonde, generalmente costituite da poche migliaia di basi, possono essere ottenute dal DNA genomico o prodotte costruendo un DNA complementare (cDNA) a specifici mRNA mediante l’uso di una transcrittasi inversa. Il cDNA differisce dal DNA genomico in quanto è privo delle sequenze introniche.
Grandi quantità di DNA genomico o di cDNA possono essere prodotte mediante clonazione, inserendo il materiale genetico, ottenuto con l’ausilio di enzimi di restrizione, in vettori (plasmidi, batteriofagi) capaci di replicarsi in batteri (ad esempio nell’Escherichia coli). Piccole sequenze di oligonucleotidi (circa 20 basi) possono essere ottenute per sintesi.
Le sonde possono essere marcate con elementi radioattivi. Nella nick translation sono introdotti dei nucleotidi marcati con 32-P in una molecola di DNA a doppio filamento. A tal fine, la molecola di DNA viene prima digerita parzialmente con una DNAasi in modo da lasciare su entrambi i filamenti delle interruzioni (nicks) in posizione casuale; queste interruzioni sono quindi utilizzate dalla DNA-polimerasi I da Escherichia coli per rimuovere e sostituire i nucleotidi non radioattivi con dei nuovi utilizzando i nucleotidi trifosfati marcati aggiunti alla miscela di reazione. Con la tecnica del random priming viene invece sintetizzata una nuova catena di DNA marcato a partire da un DNA a singolo filamento usando, come primer, una miscela di corti oligonucleotidi capaci di ibridizzare comunque con il DNA e, come enzima, la porzione della DNA-polimerasi I da Escherichia coli priva di attività esonucleasica 5’-3’ (frammento di Klenow). Con il metodo dell’end labelling, il DNA viene marcato con 32-P all’estremità 5’ mediante la reazione catalizzata dalla polinucleotide chinasi. E’ possibile evitare gli svantaggi derivanti dall’uso di tecniche di autoradiografia marcando le sonde con sostanze non radioattive: fluorofori (ad es. CyDye, Amersham), apteni (ad es. la digossigenina) rilevabili con anticorpi, sistemi utilizzanti il complesso biotina/avidina in presenza di un fluoroforo .
Nella tecnica della individuazione allelica ligasi-mediata vengono utilizzati due oligonucleotidi, uno marcato con biotina e l’altro con 32-P, che fiancheggiano un sito di mutazione e che possono essere uniti da una ligasi solo se complementari alla sequenza di DNA in esame. Il legame fra i due nucleotidi può essere dimostrato mediante cattura del prodotto di reazione con streptavidina e dosaggio della radioattività.
14.6.6d. Ibridazione in sito
La ibridazione in sito consente di ricercare la presenza e di determinare la posizione di molecole di DNA o RNA in tessuti, cellule od organuli subcellulari mediante l’uso di sonde a singolo filamento costituite da oligonucleotidi di DNA o RNA antisenso. Le sonde possono essere marcate direttamente prima dell’uso o evidenziate mediante anticorpi specifici dopo l’ibridazione del campione. Le sonde possono essere marcate con radioisotopi, fluorofori od oro colloidale e possono essere visualizzate mediante autoradiografia, microscopia ottica (Fluorescence In Situ Hybridization, FISH) od elettronica. L’utilizzazione di varianti della FISH, quali la mFISH (multicolor FISH) e lo SKY (spectral karyotyping), che utilizzano più marcatori contemporaneamente, permette di aumentare notevolmente la precisione dell’analisi e di ottenere cariotipi ad alta risoluzione (Fig. 14.3).
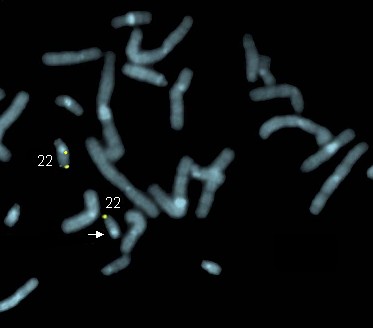
Fig. 14.3. Fluorescence In
Situ Hybridization
(FISH).
Localizzazione di un difetto associato con la sindrome di Di George
![]() .
La perdita del segnale su uno dei cromosomi 22 indica una delezione
.
La perdita del segnale su uno dei cromosomi 22 indica una delezione
![]()
14.6.6e. Ibridazione su filtro
L’ibridazione su filtro, nelle sue numerose varianti,
consiste nell’immobilizzare l’acido nucleico da
analizzare su un supporto inerte (nitrocellulosa, nylon) in modo
che le catene complementari non possano riassociarsi e restino
disponibili per la reazione di ibridazione con la sonda marcata.
La velocità e la specificità di ibridazione dipendono dalla
concentrazione, complessità e dimensioni della sonda e dalla
temperatura, forza ionica e pH del mezzo. Questa metodica può
essere adoperata con il DNA o con l’RNA e richiede che l’acido
nucleico sia denaturato ![]() .
.
La tecnica del dot blot consiste nell’applicare direttamente su filtro il campione di DNA o RNA non purificato sotto forma di una macchia puntiforme. Questa tecnica è rapida, di facile allestimento e permette di analizzare numerosi campioni sul medesimo filtro o di ottenere una valutazione semiquantitativa dell’acido nucleico ibridizzato confrontando il segnale fornito da diluizioni seriate del campione. La tecnica dell’Allele-Specific Oligonucleotide (ASO) utilizza sonde oligonucleotidiche sintetiche di circa 20 residui, una complementare al fenotipo standard e l’altra all’allele mutante con il sito di mutazione nel mezzo della regione complementare, in modo tale che, in opportune condizioni di ibridizzazione, gli oligonucleotidi si possano legare solo in caso di totale complementarietà. Nel metodo dell’ASO inverso, vengono immobilizzati gli oligonucleotidi ed il materiale da testare è utilizzato come target, permettendo di analizzare simultaneamente molti alleli (Fig. 14.4).
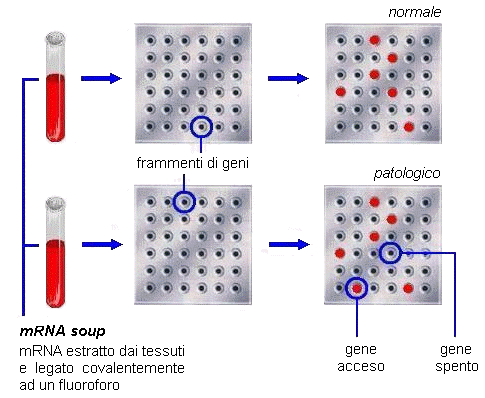
Fig. 14.4. Ibridazione su filtro mediante il metodo dell’ASO inverso. L’RNA estratto dai tessuti è stato legato ad un fluoroforo e cimentato con una serie di oligonucleotidi sintetici disposti in una matrice al fine di identificare geni espressi in modo specifico dalle cellule normali e patologiche.
La tecnica di Southern blotting consiste nel separare i frammenti di DNA (generati solitamente da una digestione con enzimi di restrizione) mediante elettroforesi in gel di agarosio e nel trasferirli su un filtro di nitrocellulosa o di nylon per capillarità. I frammenti, immobilizzati sul nuovo supporto, conservano la posizione reciproca e possono essere così ibridizzati con una sonda specifica opportunamente marcata. Nel caso si usi DNA amplificato mediante PCR, questo può essere visualizzato direttamente sotto luce UV dopo aggiunta di bromuro di etidio. Una tecnica analoga alla precedente (Northern blotting) è utilizzata per la separazione e l’analisi dell’mRNA estratto da campioni biologici al fine di studiare l’espressione di determinati geni.
14.6.6f. DNA arrays
I dispositivi di ibridazione che permettono di analizzare contemporaneamente un enorme numero di sequenze di DNA (immobilizzate su un supporto solido secondo una disposizione a matrice) sono indifferentemente chiamati DNA arrays, gene arrays, biochips o DNA chips. Il termine Gene Chips, spesso usato come sinonimo dei precedenti, si riferisce più propriamente ad un marchio depositato dalla Affymetryx Incorporated con sede a Santa Clara in California e relativo ad un dispositivo commercializzato da questa ditta.
I dispositivi prendono il nome di macroarrays se ogni singola macchia puntiforme di DNA immobilizzato ha un diametro superiore a 200-300 μm. Essi sono generalmente costituiti da matrici formate da qualche centinaio di punti, possono essere costruiti manualmente e sono analizzabili mediante l’utilizzo dei comuni lettori di gel. I dispositivi prendono il nome di microarrays (o microchips) quando contengono sul medesimo supporto una matrice con diverse migliaia di macchie puntiformi di DNA, ciascuna con un diametro inferiore a 200 μm. A differenza dei precedenti, la costruzione di questi dispositivi richiede l’uso di speciali apparecchiature robotizzate. Il supporto su cui vengono depositate le catene di DNA è generalmente costituito da una lastrina di vetro o di nylon larga qualche centimetro.
Il DNA presente sui microchips può essere costituito da oligonucleotidi formati da 20-80 basi che sono sintetizzati o direttamente in situ (on-chip) o mediante metodi convenzionali e successivamente fissati sulla matrice; questi dispositivi, tradizionalmente indicati come DNA chips, furono inizialmente sviluppati dalla Affymetryx utilizzando metodiche di fotolitografia. Una metodica alternativa, sviluppata inizialmente presso la Standford University, consiste nell’immobilizzare su lastrina delle catene di cDNA a struttura nota; i dispositivi costituiti da cDNA sono tradizionalmente indicati con il nome di DNA microarrays (Fig. 14.5).
Fig. 14.5. DNA microarray ibridizzato con un target fluorescente.
I campioni da analizzare sono preparati a partire dai tessuti biologici. Negli studi di tipizzazione genica i campioni sono costituiti da DNA genomico, mentre quando viene studiata l’espressione dei singoli geni si utilizza il mRNA estratto dalle cellule o il cDNA che deriva dal precedente per azione della trascrittasi inversa. L’acido nucleico da analizzare viene reso radioattivo o, più frequentemente, viene reso fluorescente mediante l’aggiunta di opportuni marcatori. Il materiale viene quindi cimentato sul microarray in modo da permettere l’ibridizzazione con la catena complementare di nucleotidi fissata sulla lastrina. L’acido nucleico presente nel campione viene infine identificato dalla posizione assunta nella matrice di punti mediante l’uso di un opportuno lettore robotizzato. Dall’intensità del segnale, rilevato in una specifica posizione lungo la matrice di punti, è possibile risalire alla concentrazione dell’acido nucleico nel campione biologico.
La tecnologia dei microarrays ha una vasta gamma di potenziali applicazioni in quanto permette di analizzare contemporaneamente un enorme numero di sequenze nucleotidiche. Questa tecnologia è particolarmente utile per la diagnosi di malattie dovute a mutazioni genetiche così come per identificare geni espressi in modo specifico in particolari cellule o tessuti normali o patologici al fine di consentire l’identificazione istologica del campione. Un altro importante campo di applicazione dei microarrays è nella farmacogenomica e nella tossicogenomica. Mediante questo metodo è infatti possibile correlare il profilo genetico del paziente alla risposta individuale al farmaco, identificare possibili bersagli terapeutici e desumere il meccanismo d’azione dei farmaci stessi analizzando contemporaneamente l’espressione di migliaia di geni in un singolo test.
14.6.6g. Sequenziamento del DNA
Le tecniche di Maxam e Gilbert e di Sanger per il sequenziamento del DNA si fondano sulla sintesi di una serie di molecole di DNA a filamento singolo, ognuna più lunga dell’altra di una base. Queste molecole possono essere separate mediante elettroforesi generando una scala da cui può essere letta la sequenza.
La tecnica di Maxam e Gilbert utilizza DNA marcato con 32-P in posizione 5’ e modificato chimicamente (mediante metilazione delle purine o scissione con idrazina delle pirimidine) in modo che la catena possa rompersi casualmente in un solo punto, coinvolgendo un solo tipo di base in ciascuna di quattro diverse miscele di reazione. La separazione elettroforetica dei frammenti su un unico gel permette di stabilire la loro lunghezza e di determinare la sequenza del DNA dal confronto reciproco delle bande radioattive.
La tecnica di Sanger utilizza una DNA-polimerasi per sintetizzare una copia complementare del DNA bersaglio in presenza di un 2’,3’-dideossinucleotide trifosfato, a partire da un primer sintetico allineato vicino alla regione da analizzare (Fig. 14.6).
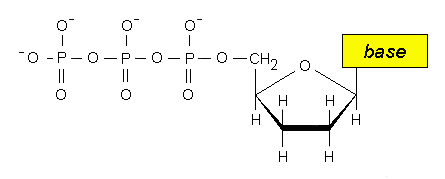
Fig. 14.6. 2’, 3’-Dideossinucleotide trifosfato.
La sintesi viene realizzata in quattro diversi mezzi di incubazione, ognuno contenente i quattro 2’-deossinucleotidi trifosfati (uno dei quali marcato radioattivamente) e basse concentrazioni di uno solo dei quattro analoghi privi anche del gruppo ossidrile in 3’. Poiché la sintesi del DNA si blocca quando l’enzima incorpora l’analogo del substrato, nel corso della reazione verranno prodotte delle popolazioni di DNA marcato di diversa lunghezza che potranno essere confrontate tra loro elettroforeticamente come già descritto precedentemente (Fig. 14.7).
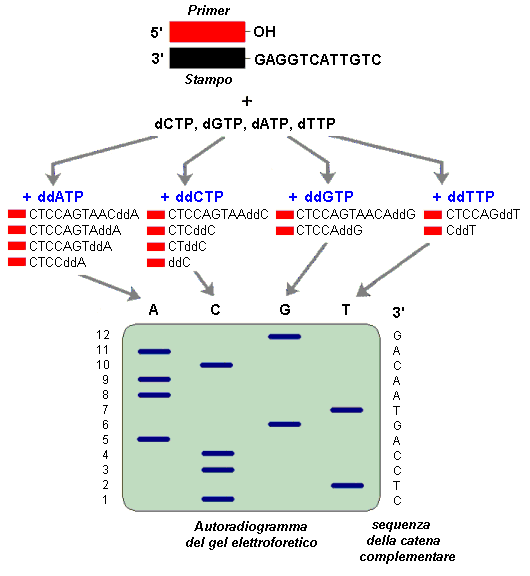
Fig. 14.7. Rappresentazione schematica del metodo di Sanger per il sequenziamento del DNA.
| aggiornamento: 26/07/11 |