|
|
Costantino Salerno |
|
|
Appunti di Biochimica Clinica |
|
|
Il sangue è un sistema bifasico costituito da una parte liquida, il plasma, e da una parte cellulare, formata a sua volta da tre principali componenti, gli eritrociti, i leucociti e le piastrine. In particolari condizioni, il plasma può andare incontro a coagulazione per trasformazione del fibrinogeno idrosolubile in un gel costituito da un reticolo di fibrina. Dopo la formazione del coagulo, la componente plasmatica che rimane ancora liquida viene definita siero.
Il volume totale del sangue circolante, o volemia, dipende dall’età, dal
sesso, dal peso e dalla superficie corporea di ciascun individuo
![]() . In condizioni
fisiologiche, la volemia è mantenuta costante nelle sue componenti cellulare e
plasmatica attraverso complessi meccanismi di regolazione. Il rapporto
percentuale della componente cellulare eritrocitaria rispetto al sangue intero viene indicato come valore di ematocrito.
. In condizioni
fisiologiche, la volemia è mantenuta costante nelle sue componenti cellulare e
plasmatica attraverso complessi meccanismi di regolazione. Il rapporto
percentuale della componente cellulare eritrocitaria rispetto al sangue intero viene indicato come valore di ematocrito.
7.1. ESAME EMOCROMOCITOMETRICO
L’esame emocromocitometrico consiste nella valutazione di una serie di parametri che riguardano le principali popolazioni cellulari presenti nel sangue periferico. Questi parametri comprendono la concentrazione dell’emoglobina (HGB), l’ematocrito (PCV, HCT), il conteggio degli eritrociti (RBC), dei leucociti (WBC) e delle piastrine (PLP), e la formula leucocitaria, intesa come rapporto percentuale fra le diverse componenti della serie bianca. Vengono inoltre calcolati alcuni indici cellulari, come il diametro medio (MCD) e il volume medio (MCV) degli eritrociti, il contenuto medio (MCH) e la concentrazione media (MCHC, CHCM) dell’emoglobina nelle singole cellule, il volume medio delle piastrine (MPV) e l’ematocrito piastrinico (PCT). Sono infine determinati alcuni parametri di dispersione dei dati, come la deviazione standard o il coefficiente di variazione del volume degli eritrociti (RDW) e del loro contenuto in emoglobina (HDW), nonché la deviazione standard o il coefficiente di variazione del volume delle piastrine (PDW). Un elenco dei principali parametri presi in considerazione nell’esame emocromocitometrico è riportato in Tab. 7.I.
Tab. 7.I. Valori di riferimento dei principali parametri ematici
| Parametro | valore | unità | |||||
| maschi | femmine | ||||||
| RBC | red blood cell count |
4,5 - 5,8 |
4,2 - 5,2 | cellule x 106/μL | |||
| MCV | mean corpuscolar volume (PCV/RBC) | 82 - 92 | f L | ||||
| RDW | red distribution width | 11,6 - 14,6 | % | ||||
| MCD | mean corpuscolar diameter | 6,7 - 7,7 | μm | ||||
| PCV | packed cell volume | 40 - 52 | 37 - 47 | % (*) | |||
| HCT | haematocrit | ||||||
| HGB | hemoglobin concentration | 13 - 17 | 12 - 16 | g /dL | |||
| MCH | mean corpuscolar hemoglobin (HGB/RBC) | 27 - 31 | pg | ||||
| MCHC | mean corpuscolar hemoglobin concentration (HGB/PCV) | 32 - 36 | g /dL (**) | ||||
| CHCM | cellular hemoglobin concentration mean | ||||||
| HDW | hemoglobin distribution width | 2,2 - 3,5 | g /dL | ||||
| PLT | platelet count | 150 - 300 | cellule x 103/μL | ||||
| MPV | mean platelet volume | 6,4 - 9,7 | f L | ||||
| PDW | platelet distribution width | 44 - 56 | % | ||||
| PCT | platelet haematocrit | 0,16 - 0,24 | % | ||||
| WBC | white blood cell count | 4 - 10 | cellule x 103/μL | ||||
| NEUT | neutrofili | 40 - 75 | % | ||||
| LINF | linfociti | 20 - 45 | % | ||||
| MONO | monociti | 2 - 10 | % | ||||
| EOS | eosinofili | 1 - 6 | % | ||||
| BASO | basofili | < 1 | % | ||||
(*) La sigla PCV si riferisce a misure effettuate mediante
centrifugazione del campione, mentre la sigla HCT si riferisce a stime
ottenute mediante apparecchiature automatiche.
(**) La sigla CHCM si riferisce a misure
dirette della concentrazione dell’emoglobina corpuscolare media mediante
light scattering, mentre la sigla MCHC si riferisce a stime
ottenute rapportando al valore di ematocrito la concentrazione dell’emoglobina ottenuta mediante
misure colorimetriche.
7.1.1. Metodi di conta manuale
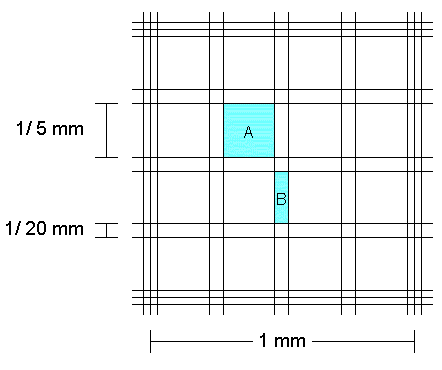
Il conteggio manuale degli elementi figurati viene effettuato utilizzando apposite camere di conta nelle quali viene immessa la soluzione da esaminare. Per la conta dei globuli rossi si utilizza del sangue diluito 1:200 in soluzione isotonica; per la conta dei globuli bianchi si utilizza del sangue diluito 1:100 in una soluzione acquosa di acido acetico (ad esempio il liquido di Türk) in modo da emolizzare solo i globuli rossi. La camera di conta di Bürker consiste in una robusta lastra quadrangolare di vetro con in mezzo 2 camere di conteggio su cui sono incisi due fini reticoli quadrati (Fig. 7.1). Le camere sono ricavate nella lastra in modo da delimitare uno spazio di 0,1 mm di spessore quando viene applicato su di esse un vetrino coprioggetto. La perfetta aderenza del vetrino è assicurata dalla comparsa di anelli iridescenti di Newton ai propri margini dopo l’applicazione di due mollette di fissaggio. La conta viene effettuata al microscopio ottico adoperando un obiettivo a secco di 40 X e oculari da 6 a 10 diametri. La conta manuale dei globuli rossi può dare luogo a risultati soddisfacenti solo se è eseguita con molta cura, ma è generalmente inadatta nella routine di laboratorio perché troppo lenta e notevolmente imprecisa. Anche la conta manuale dei globuli bianchi è imprecisa; ciò è tuttavia meno importante dal punto di vista pratico in quanto le alterazioni che hanno una rilevanza clinica sono il più delle volte sufficientemente pronunciate da poter essere osservate anche con questa tecnica.
Il conteggio manuale delle piastrine può essere effettuato sul sangue intero o su plasma arricchito. Se sono presenti piastrine molto grandi è preferibile il primo metodo per evitare di perdere delle piastrine durante l’arricchimento; se il numero di piastrine è basso è invece preferibile arricchire il plasma mediante centrifugazione. Piastrine molto grandi possono essere distinte dai globuli rossi in base alla loro diversa morfologia. Si possono ottenere risultati più accurati se si eliminano i globuli rossi, sottoponendoli a lisi mediante ossalato d’ammonio. Le piastrine possono essere facilmente visualizzate in contrasto di fase o dopo colorazione con brilliant cresyl blue. La conta manuale è tuttavia generalmente imprecisa e particolarmente laboriosa.
La conta differenziale dei globuli bianchi può essere eseguita manualmente al microscopio ottico su uno striscio di sangue opportunamente colorato. I leucociti sono classificati in un numero discreto di categorie (generalmente 5 o 6 se viene fatta una distinzione fra i neutrofili a nucleo segmentato e quelli a nucleo non segmentato). I globuli rossi nucleati (NRBC, nucleated red blood cells) costituiscono una ulteriore categoria di cellule che può essere contata a parte o essere inclusa nella conta dei leucociti ed espressa come percentuale del numero totale di cellule nucleate (TNCC, total nucleated cell count). La conta differenziale manuale è soggetta ad errori derivanti da una cattiva distribuzione delle cellule sul vetrino o da una cattiva interpretazione dei risultati.
Le cellule non sono ugualmente distribuite lungo lo striscio di sangue: la coda dello striscio contiene più neutrofili e meno linfociti, mentre i monociti sono distribuiti più uniformemente; le grosse cellule immature sono presenti preferibilmente ai lati e nelle zone distali dello striscio. Risultati peggiori si hanno quando lo striscio è troppo sottile o è stato eseguito con una cattiva angolazione. In pratica, l’imprecisione della conta manuale è talmente elevata che un piccolo grado di inaccuratezza nella distribuzione delle cellule è generalmente privo di conseguenze. Tuttavia, se sono presenti degli aggregati di cellule, la cattiva distribuzione è così grande da rendere impossibile una conta differenziale dei leucociti. Una cattiva interpretazione dei risultati è infrequente in mani esperte. La descrizione dettagliata delle caratteristiche morfologiche delle singole cellule esula dalla presente trattazione.
7.1.1a. Determinazione dell’ematocrito
L’ematocrito può essere valutato manualmente utilizzando dei capillari di vetro (generalmente lunghi 75 mm e con un diametro interno di 1,5 mm) che vengono riempiti con il sangue, chiusi ad una estremità al calore o con del mastice e centrifugati per 5 - 10 minuti in appositi rotori. Il volume dei globuli rossi impaccati (PCV, packed cell volume) è valutato su una scala graduata escludendo dalla misura il plasma e lo strato formato dai globuli bianchi e dalle piastrine (buffy coat).
La misura è poco precisa e poco accurata. La lettura sulla scala graduata può essere resa difficile dalle piccole dimensioni del tubo; il fondo del tubo è generalmente concavo se è stato chiuso al calore, mentre può essere convesso se è stato chiuso con il mastice; le cellule possono aver subìto una contrazione di volume a seguito dell’aggiunta dell’anticoagulante (l’ematocrito risulta inferiore di circa il 2% se viene usato l’EDTA come sale tripotassico al posto del sale bipotassico; vedi Par. 7.1.6); una parte del plasma (valutabile attorno al 2 - 3%) rimane intrappolata fra le cellule determinando una sovrastima del volume dei globuli rossi.
7.1.2. Apparecchiature automatiche
Le moderne apparecchiature per l’analisi automatica dei campioni di sangue sono in grado di determinare tutta una serie di parametri riguardanti i globuli rossi, i globuli bianchi e le piastrine. Alcune apparecchiature sono anche in grado di identificare da sole il campione da esaminare attraverso la lettura di un codice a barre, di controllare se esso ha un volume adeguato ed è privo di coaguli, di prelevare automaticamente il sangue dalla provetta e di preparare strisci di sangue per eventuali ulteriori esami. Le apparecchiature automatiche sono progettate per fornire conte accurate e precise su campioni di sangue normali o con lievi alterazioni numeriche rispetto al normale e per allertare l’operatore quando il campione presenta caratteristiche inusuali (presenza di cellule blastiche, granulociti immaturi, piastrine giganti o aggregati piastrinici) tali da richiedere uno studio più accurato al microscopio ottico.
Tutte le determinazioni (ad eccezione di quella relativa all’emoglobina totale, vedi Par. 7.2.2) si basano generalmente sulla misura del numero e delle dimensioni degli elementi figurati. Le apparecchiature automatiche hanno normalmente almeno due canali. Il primo canale è utilizzato per la conta e la misura delle dimensioni dei globuli rossi e delle piastrine in un campione opportunamente diluito. Nel secondo canale è aggiunto al diluente un agente litico in modo da lasciare intatti per la conta solo i globuli bianchi. Quest’ultimo canale può essere utilizzato anche per calcolare la concentrazione dell’emoglobina, generalmente attraverso la determinazione colorimetrica della cianometemoglobina. In alternativa, la concentrazione dell’emoglobina può essere determinata con il metodo che utilizza il laurilsolfato di sodio. Ulteriori canali sono necessari per una conta differenziale dei globuli bianchi e dei reticoliti.
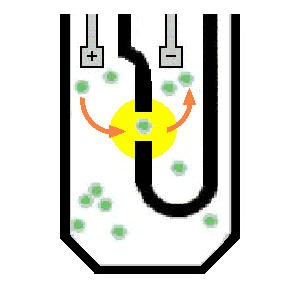
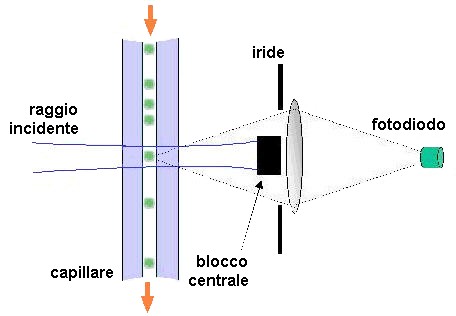
La conta degli elementi figurati e la determinazione della loro dimensione vengono effettuate mediante misure di impedenza elettrica o di luce diffusa (light scattering; Fig. 7.2). I contatori ad impedenza, sviluppati su un progetto iniziale di Wallace Coulter attorno al 1950, sfruttano la proprietà delle cellule del sangue di essere dei pessimi conduttori elettrici. Per questo motivo, quando le cellule opportunamente diluite sono fatte passare ad una ad una per un piccolo foro attraverso il quale è stata applicata una corrente elettrica, si osserva un aumento di impedenza che è proporzionale al volume delle singole cellule. Queste possono essere così contate e misurate in relazione al loro volume sulla base dell’impulso elettrico generato. Bisogna tenere però presente che diversi fattori possono disturbare il segnale. Le cellule con maggiore deformabilità, che assumono una forma allungata quando passano attraverso il foro, e le cellule che ricircolano ai margini del foro stesso generano un segnale più piccolo e sembrano perciò di minori dimensioni, mentre le cellule a struttura rigida o che non passano al centro del foro possono sembrare più grandi. Inoltre, quando più cellule passano simultaneamente attraverso il foro, esse vengono contate come una singola cellula. Si può tuttavia ovviare almeno parzialmente a questi errori regolando adeguatamente il flusso delle cellule o mediante correzioni elettroniche. I contatori a luce diffusa, messi a punto in anni più recenti, utilizzano la luce bianca (nel caso dei leucociti) o la luce laser (nel caso dei globuli rossi e delle piastrine) per contare gli elementi figurati e determinarne il volume in base all’intensità della luce diffusa, osservata ad un angolo compreso fra 1° e 3° rispetto al raggio incidente (forward light scatter).
|
|
|
|
Contatore Coulter ad impedenza |
Contatore a diffusione luminosa |
|
|
|
7.1.2a. Conta dei globuli rossi e delle piastrine
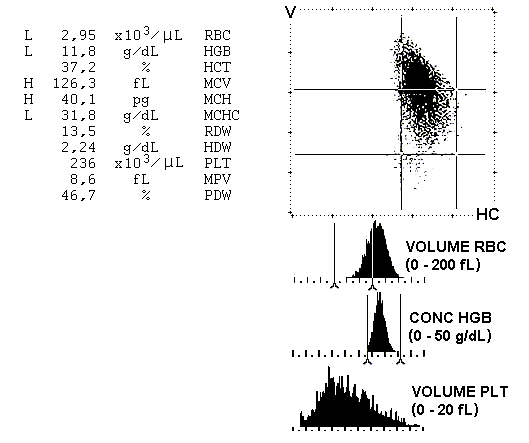
I globuli rossi e le piastrine sono generalmente contati sul medesimo canale e differenziati in base alle dimensioni (le piastrine hanno normalmente un volume inferiore a 20 fL). Le distribuzioni di frequenza dei rispettivi volumi (generalmente riportate su appositi istogrammi) permettono di ricavare i valori di MCV e RDW per i globuli rossi e di MPV e PDW per le piastrine. Il valore di PCV è calcolato dai valori di MCV e RBC. Il valore di MCH è calcolato dal valore di RBC e dal valore di HGB (determinato separatamente mediante misure colorimetriche). Il valore di MCHC è calcolato dai valori di RBC, HGB e MCV.
Utilizzando rivelatori capaci di misurare la luce diffusa a due angolazioni diverse rispetto alla luce incidente (2°-3° e 5°-15°), è possibile calcolare contemporaneamente sia il volume delle singole cellule sia la concentrazione di emoglobina presente in esse. Ciò permette di determinare direttamente il valore di CHCM ed i parametri da esso derivabili senza dover misurare separatamente la concentrazione dell’emoglobina mediante metodi colorimetrici. La distribuzione di frequenza dei valori di concentrazione cellulare dell’emoglobina permette di valutare il valore di HDW (Fig. 7.3).
 |
|
|
La conta automatica dei reticolociti viene effettuata misurando la fluorescenza, l’assorbimento o la diffusione luminosa delle cellule che sono state cimentate con dei fluorofori o dei cromofori (new methylene blue, oxazin 750) capaci di legarsi all’RNA. Poiché queste sostanze si legano anche al DNA delle cellule nucleate, è necessario escludere i segnali che superano determinati valori di soglia. L’intensità del segnale è proporzionale all’RNA presente nei reticolociti ed è perciò un indice della loro immaturità (Fig. 7.4). La misura dipende comunque dalle condizioni sperimentali impiegate (tipo di sonda utilizzata, tempi di reazione, temperatura) e per questo motivo i valori di riferimento presentano forti variazioni da apparecchio ad apparecchio. Il numero di reticolociti in un campione di sangue tende inoltre a diminuire nel tempo (cala del 5% dopo un giorno di incubazione a temperatura d’ambiente) a causa della progressiva maturazione delle cellule.
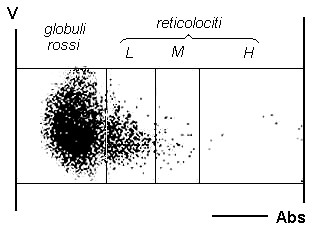
7.1.2b. Conta differenziale dei globuli bianchi
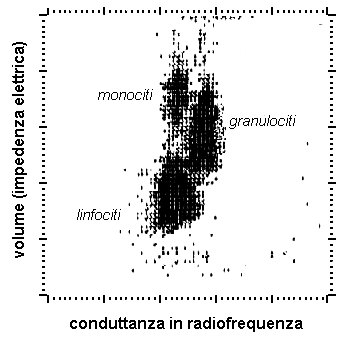
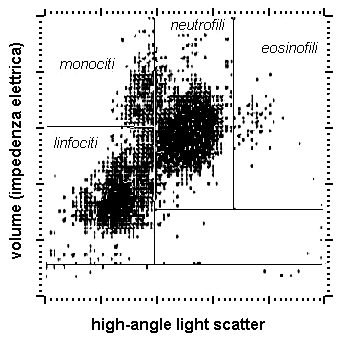
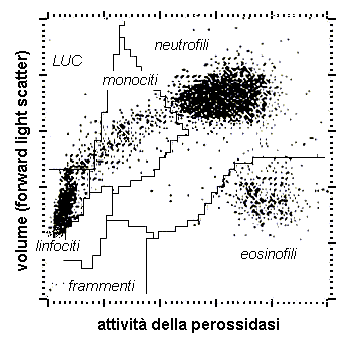
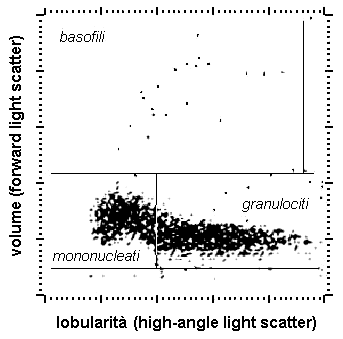
La conta differenziale dei globuli bianchi viene effettuata prendendo in considerazione contemporaneamente due o più parametri discriminanti (uno di essi è quasi sempre il volume) in modo da poter suddividere in un grafico a più dimensioni tutta la popolazione cellulare in diversi sottogruppi (Fig. 7.5).
Misure di conduttanza o di capacitanza in radiofrequenza e misure di diffusione luminosa con una angolazione di 7°-11° (narrow-angle light scatter) permettono di ottenere segnali che dipendono dalla complessità della cellula e dalla sua struttura interna (rapporto nucleo/citoplasma, densità del nucleo, granulosità del citoplasma). Segnali ottenuti a più ampie angolazioni (high-angle light scatter) dipendono dalla densità e dalla lobulazione del nucleo, valutabile attraverso un indice (LI, lobularity index) che assume valori decrescenti passando dai neutrofili alle cellule mononucleate, ai granulociti immaturi ed ai blasti. I granulociti eosinofili sono differenziabili dai neutrofili per la loro capacità di depolarizzare la luce diffusa con una angolazione di 70°-100° (depolarizaed orthogonal light scatter).
Il DNA e l’RNA delle cellule può essere marcato con un fluoroforo (polymethine fluorescent dye). I granuli citoplasmatici possono essere colorati e valutati mediante tecniche di assorbimento ottico (il chlorazol black si lega fortemente ai granuli eosinofili, in misura intermedia ai neutrofili, debolmente ai monociti, mentre non colora i linfociti). L’attività della perossidasi dei neutrofili, eosinofili e monociti può essere dosata utilizzando il 4-cloro-1-naftolo come substrato (dalla reazione si sviluppa un colore nero misurabile mediante un fotometro). Quest’ultima tecnica permette di determinare un indice di attività perossidasica (MPXI, mean peroxidase activity index) nonché di differenziare le cellule con attività perossidasica da quelle che ne sono prive, come i granulociti basofili ed i linfociti normali ed atipici (LUC, large unstained cells).
Un’altra possibilità è di sfruttare la differente resistenza alla lisi dei vari tipi di cellule in modo da isolare una serie dalle altre. I granulociti eosinofili e basofili sono più resistenti agli agenti litici rispettivamente in ambiente alcalino ed acido. Dopo aver sottoposto il sangue ad una lisi parziale in condizioni opportune, è possibile perciò contare gli eosinofili ed i basofili separatamente dalle altre cellule e classificarli in base al loro volume.
 |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
7.1.3. Alterazioni della serie rossa
Le alterazioni degli eritrociti possono riguardare il loro numero, forma, volume e contenuto in emoglobina. Quando in uno striscio di sangue non vi sono grandi variazioni nelle caratteristiche di grandezza, forma e colorabilità delle cellule si parla di normocitosi. La presenza di una variabilità nelle dimensioni degli eritrociti è definita anisocitosi, mentre la presenza di una variabilità nella forma è definita poichilocitosi. Alcune alterazioni degli eritrociti hanno delle particolari denominazioni: si può parlare così di sferocitosi (perdita della forma a disco biconcavo), elissocitosi (cellule a forma ovale), leptocitosi (cellule con forma a bersaglio per la presenza dell’emoglobina in una piccola zona centrale), stomatocitosi (presenza di una area pallida centrale allungata a fessura), acantocitosi (superficie irregolarmente spinosa), echinocitosi (superficie regolarmente spinosa), schistocitosi (presenza di frammenti cellulari in circolo). Dal punto di vista classificativo, è utile distinguere le alterazioni della serie rossa in anemie ed eritrocitosi.
7.1.3a. Anemie
Sono definite "anemie" quelle condizioni morbose caratterizzate da un diminuito contenuto di emoglobina per unità di volume di sangue circolante (HGB). Dal punto di vista generale, le anemie si dividono in macrocitiche, normocitiche e microcitiche a seconda che il volume medio degli eritrociti (MCV) sia inferiore alla norma, nella norma o superiore alla norma. Se si tiene conto della concentrazione dell’emoglobina nei globuli rossi (MCHC, CHCM), le anemie si classificano in ipocromiche, normocromiche, e ipercromiche a seconda che questo parametro sia inferiore alla norma, nella norma o superiore alla norma. Per quanto riguarda la patogenesi, le anemie possono derivare da una perdita di sangue acuta o cronica, da un deficit di eritropoiesi o da una eccessiva emolisi dei globuli rossi (Tab. 7.II).
|
Tab. 7.II. Classificazione etiologica delle anemie |
||
| Anemie postemorragie | ||
| anemie acute | ||
| anemie croniche | ||
| Anemie da deficit di eritropoiesi | ||
| anemie normocromiche-normocitiche | ||
| anemia ipoproliferativa | ||
| panipoplasia midollare | ||
| aplasia pura della serie rossa | ||
| mielodisplasia | ||
| mieloftisi | ||
| anemie microcitiche | ||
| deficit di ferro | ||
| errori nella biosintesi della globina | ||
| malattie croniche | ||
| anemie macrocitiche | ||
| anemie megaloblastiche | ||
| anemie non-megaloblastiche | ||
| Anemie emolitiche | ||
| da fattori estrinseci | ||
| malattie autoimmuni | ||
| agenti infettivi | ||
| ipersplenismo | ||
| trattamenti dialitici | ||
| traumi | ||
| da deficit di fattori intrinseci | ||
| difetti degli enzimi eritrocitari | ||
| difetti della membrana eritrocitaria | ||
| difetti dell’emoglobina | ||
(1) L’anemia postemorragica acuta è dovuta ad una massiccia ed immediata perdita di sangue. Durante e immediatamente dopo il fenomeno emorragico, i valori di RBC, HGB e HCT possono aumentare a seguito della vasocostrizione, ma nelle ore immediatamente successive, a causa dell’emodiluizione, si assiste ad una rapida caduta di questi parametri con un conseguente quadro di anemia normocitica. In queste prime ore si può inoltre osservare un aumento in circolo dei granulociti neutrofili e delle piastrine. Diversi giorni dopo può aumentare il numero di reticolociti (reticolocitosi postemorragica) e si possono osservare occasionalmente in circolo normoblasti e cellule immature della serie bianca. Nel caso di una perdita cronica di piccole o moderate quantità di sangue dal tratto gastrointestinale (ulcere peptiche, emorroidi), urinario o genitale, il quadro è quello di una anemia microcitica ed i dati laboratoristici sono simili a quelli osservabili in caso di carenza di ferro. Una anemia ipocromica microcitica è anche osservabile in caso di perdite croniche di sangue per deficit di vitamina C; in altri casi questo deficit vitaminico dà luogo ad una anemia normocitica, se isolato, o ad una anemia macrocitica, se associato a carenza di acido folico.
(2) Un deficit dell’eritropoiesi può portare ad una anemia normocromica-normocitica, ad una anemia microcitica o ad una anemia macrocitica. Poiché la vita media dei globuli rossi nel sangue è di 120 giorni, un blocco anche completo dell’eritropoiesi porta ad una diminuzione di non più di circa il 10% di cellule alla settimana. Il deficit di eritropoiesi è inoltre generalmente accompagnato a reticolopenia.
Un quadro di anemia normocromica-normocitica fa generalmente pensare ad un
meccanismo ipoproliferativo o ipoplastico. Questo può essere associato a
carenza di eritropoietina
![]() (anemia ipoproliferativa da malattie renali, da
deprivazione proteica, da stati ipometabolici), a panipoplasia midollare (per
avvelenamenti, radiazioni ionizzanti, infezioni acute, cause genetiche
(anemia ipoproliferativa da malattie renali, da
deprivazione proteica, da stati ipometabolici), a panipoplasia midollare (per
avvelenamenti, radiazioni ionizzanti, infezioni acute, cause genetiche
![]() ), ad aplasia pura
della serie rossa (per infezioni da parvovirus o di altro tipo, avvelenamenti,
deficit di riboflavina, disordini emolitici, timoma, leucemia linfoide cronica), a mielodisplasia
), ad aplasia pura
della serie rossa (per infezioni da parvovirus o di altro tipo, avvelenamenti,
deficit di riboflavina, disordini emolitici, timoma, leucemia linfoide cronica), a mielodisplasia
![]() o a mieloftisi
(per infiltrazione dello spazio midollare con cellule non-ematopoietiche
generalmente di natura neoplastica). La mieloftisi è generalmente accompagnata
ad anosocitosi, poichilocitosi e presenza di emazie nucleate in circolo.
o a mieloftisi
(per infiltrazione dello spazio midollare con cellule non-ematopoietiche
generalmente di natura neoplastica). La mieloftisi è generalmente accompagnata
ad anosocitosi, poichilocitosi e presenza di emazie nucleate in circolo.
Un quadro microcitemico suggerisce un difetto nella sintesi dell’eme o della
globina (deficit di ferro, talassemie o altri errori nella biosintesi
della globina, malattie croniche). Le alterazioni
iniziali possono essere minime e può coesistere una ipocromia di vario grado. Le
forme più comuni di anemia sono dovute a carenza di ferro (causata generalmente
da perdite croniche di sangue, più raramente da emoglobinuria o emosiderinuria o
da una sindrome da malassorbimento). Molto più rare sono le anemie dovute a difetti
di trasporto (atransferritinemia; vedi
Par. 8.1.5) o di utilizzazione del ferro (emoglobinopatie,
anemie sideroblastiche o mielodisplastiche). I più comuni esami di laboratorio
riguardano la determinazione su campioni di sangue della ferritina (vedi
Par. 8.1.6), dei
recettori solubili della transferrina, del ferro e della capacità legante il
ferro. Le anemie in corso di malattie croniche (neoplasie,
infezioni croniche, processi infiammatori cronici come ad esempio l’artrite reumatoide
![]() ) sono un’altra forma molto comune di anemia. In questi casi,
tuttavia, poiché la ferritinemia aumenta nel corso di neoplasie o di reazioni di
fase acuta, la specificità di questo indice è minore ed è preferibile basarsi
nella diagnosi sui valori della ferritina eritrocitaria o sulla determinazione
dei recettori solubili della transferrina. Le anemie microcitiche dovute ad emoglobinopatie
sono trattate nel Par. 7.2.1.
) sono un’altra forma molto comune di anemia. In questi casi,
tuttavia, poiché la ferritinemia aumenta nel corso di neoplasie o di reazioni di
fase acuta, la specificità di questo indice è minore ed è preferibile basarsi
nella diagnosi sui valori della ferritina eritrocitaria o sulla determinazione
dei recettori solubili della transferrina. Le anemie microcitiche dovute ad emoglobinopatie
sono trattate nel Par. 7.2.1.
Le anemie macrocitiche sono dovute a un difetto nella sintesi del DNA. La
causa più comune è una carenza di vitamina B12 o di acido folico
![]() o l’uso di farmaci
antineoplastici. La carenza di vitamina B12 è generalmente dovuta a
mancata secrezione del fattore intrinseco (anemia perniciosa) o, più raramente,
ad un inadeguato apporto vitaminico, ad un aumentato consumo della vitamina da
parte di batteri o vermi intestinali, ad alterazioni congenite o resezioni
chirurgiche del tratto intestinale, a sindromi da malassorbimento o uso di
farmaci (chelanti del calcio, aminosalicilati, biguanidi). L’anemia è
caratterizzata da macro-ovalocitosi, anisocitosi e poichilocitosi. Di comune
riscontro è la presenza di frammenti di nucleo negli eritrociti (corpi di
Howell-Jolly). I granulociti mostrano una precoce ipersegmentazione, mentre solo
più tardi compare una neutropenia. In circa la metà dei casi si osserva
trombocitopenia con piastrine di dimensioni e forma bizzarre. Istologicamente,
il midollo si presenta costituito in assoluta prevalenza (dal 10 al 25% delle
cellule nucleate) da elementi atipici della serie rossa, definiti megaloblasti.
Forme di anemia macrocitica non-megaloblastica si possono invece osservare
in pazienti
affetti da mielodisplasia, malattie epatiche croniche o
alcolismo cronico.
o l’uso di farmaci
antineoplastici. La carenza di vitamina B12 è generalmente dovuta a
mancata secrezione del fattore intrinseco (anemia perniciosa) o, più raramente,
ad un inadeguato apporto vitaminico, ad un aumentato consumo della vitamina da
parte di batteri o vermi intestinali, ad alterazioni congenite o resezioni
chirurgiche del tratto intestinale, a sindromi da malassorbimento o uso di
farmaci (chelanti del calcio, aminosalicilati, biguanidi). L’anemia è
caratterizzata da macro-ovalocitosi, anisocitosi e poichilocitosi. Di comune
riscontro è la presenza di frammenti di nucleo negli eritrociti (corpi di
Howell-Jolly). I granulociti mostrano una precoce ipersegmentazione, mentre solo
più tardi compare una neutropenia. In circa la metà dei casi si osserva
trombocitopenia con piastrine di dimensioni e forma bizzarre. Istologicamente,
il midollo si presenta costituito in assoluta prevalenza (dal 10 al 25% delle
cellule nucleate) da elementi atipici della serie rossa, definiti megaloblasti.
Forme di anemia macrocitica non-megaloblastica si possono invece osservare
in pazienti
affetti da mielodisplasia, malattie epatiche croniche o
alcolismo cronico.
(3) L’emolisi è conseguente ad una riduzione della durata di vita degli eritrociti. Il midollo osseo può rispondere aumentando l’eritropoiesi (anemia emolitica compensata), ma, quando questa non riesce a compensare le perdite, l’anemia si manifesta clinicamente. Quando il catabolismo dell’eme supera la capacità funzionale del fegato, si ha la comparsa dell’ittero documentabile in laboratorio dall’aumento della bilirubina indiretta nel sangue, della stercobilina nelle feci e della urobilinogeno nelle urine (vedi Par. 5.2). Comune è il riscontro di un aumento della lattico deidrogenasi plasmatica (Par. 9.1.1). Nel sangue periferico possono essere evidenziati sferociti e frammenti cellulari. L’aumento dell’eritropoiesi può determinare una reticolocitosi; questa tuttavia manca nel corso delle crisi aplastiche, caratterizzate da un temporaneo deficit dell’eritropoiesi, che possono manifestarsi specialmente a seguito di infezioni virali (parvovirus).
Generalmente l’emolisi è
extravasale ed avviene nelle cellule macrofagiche della milza, del fegato e del
midollo osseo. Più rari sono i casi di emolisi intravasale che danno luogo ad
emoglobinuria, quando l’emoglobina supera la capacità legante dell’aptoglobina
plasmatica (vedi Par. 8.1.3). L’emoglobina può essere riassorbita
dalle cellule tubolari renali, trasformata in emosiderina, riutilizzata in parte, o eliminata con le urine (emosiderinuria). L’emolisi può
essere dovuta a problemi estrinseci ai globuli rossi (incompatibilità
meterno-fetale
![]() , malattie autoimmuni,
agenti infettivi, ipersplenismo, trattamenti dialitici, traumi meccanici) o a
difetti intrinseci dei globuli rossi (difetti degli enzimi o della membrana eritrocitaria,
difetti dell’emoglobina).
, malattie autoimmuni,
agenti infettivi, ipersplenismo, trattamenti dialitici, traumi meccanici) o a
difetti intrinseci dei globuli rossi (difetti degli enzimi o della membrana eritrocitaria,
difetti dell’emoglobina).
Le anemie emolitiche autoimmuni sono diagnosticabili mediante il test di
Coombs diretto (consistente nel provocare l’agglutinazione di una preparazione
di emazie lavate del paziente mediante aggiunta di siero antiglobuline) o
indiretto (consistente nel provocare l’agglutinazione di una preparazione di emazie normali per aggiunta del siero del paziente). In base alla reattività
degli anticorpi a diverse temperature, queste anemie vengono distinte in anemia
emolitica da anticorpi caldi (le più frequenti), anemia emolitica da anticorpi
freddi ed emoglobinuria parossistica a frigore (o sindrome di Donath-Landsteiner)
![]() . Un’anemia
emolitica causata da fattori del complemento è l’emoglobinuria parossistica
notturna (o sindrome di Marchiafava-Micheli), un raro disordine dovuto ad una
ipersensibilità alla componente C3 per un difetto delle proteine di membrana del
globulo rosso; per la diagnosi si utilizza il test di emolisi acida di Ham che
consiste nell’incubare i globuli rossi in HCl diluito.
. Un’anemia
emolitica causata da fattori del complemento è l’emoglobinuria parossistica
notturna (o sindrome di Marchiafava-Micheli), un raro disordine dovuto ad una
ipersensibilità alla componente C3 per un difetto delle proteine di membrana del
globulo rosso; per la diagnosi si utilizza il test di emolisi acida di Ham che
consiste nell’incubare i globuli rossi in HCl diluito.
Alcuni agenti infettivi possono indurre anemia attraverso la produzione di tossine (Clostridium
perfrigens, streptococco α- o β-emolitico, meningococco) o distruggendo direttamente le emazie
(Plasmodio, Bartonella). Un’ulteriore causa di anemia è l’ipersplenismo secondario a
splenomegalia congestizia (cirrosi epatica, trombosi della vena porta o della
vena splenica), a malattie mielo- o linfoproliferative, a malattie da accumulo
(ad esempio il morbo di Gaucher
![]() ), a connettiviti o a malattie infettive
(malaria, kala-azar, etc.). Anche i traumi meccanici a cui sono sottoposte le
cellule ematiche possono causare emolisi. Essi possono originare all’esterno dei
vasi (emoglobinuria del marciatore o del lottatore), all’interno delle cavità
cardiache (stenosi aortiche calcificate, protesi valvolari), nelle arteriole
(ipertensione maligna) o in seguito a trattamenti emodialitici..
), a connettiviti o a malattie infettive
(malaria, kala-azar, etc.). Anche i traumi meccanici a cui sono sottoposte le
cellule ematiche possono causare emolisi. Essi possono originare all’esterno dei
vasi (emoglobinuria del marciatore o del lottatore), all’interno delle cavità
cardiache (stenosi aortiche calcificate, protesi valvolari), nelle arteriole
(ipertensione maligna) o in seguito a trattamenti emodialitici..
Una alterazione congenita di uno degli enzimi eritrocitari coinvolti nella glicolisi anaerobia, nella via dell’esosomonofosato, nel metabolismo del glutatione o nelle vie di interconversione dei nucleotidi può costituire una causa intrinseca di emolisi. I deficit di piruvato chinasi e di glucosio-6-fosfato deidrogenasi sono le cause metaboliche più frequenti di anemia emolitica. Più rari sono i difetti congeniti degli altri enzimi della glicolisi (esochinasi, glucosofosfato isomerasi, fosfofruttochinasi, aldolasi, triosofosfato isomerasi, fosfoglicerato chinasi, bisfosfoglicerato mutasi) e del metabolismo del glutatione (glutatione sintetasi, glutatione reduttasi, g-glutamilcisteina sintetasi). Le alterazioni delle vie di interconversione dei nucleotidi che danno luogo ad anemia emolitica sono una sovrapproduzione di adenosina deaminasi ed un deficit di pirimidina nucleotidasi. Tutte queste forme di anemia si manifestano come malattie ereditarie autosomiche recessive, ad eccezione dei deficit della glucosio-6-fosfato deidrogenasi e della fosfoglicerato chinasi, che sono legati al cromosoma X, della sovrapproduzione di adenosina deaminasi, che è autosomica dominante, e del deficit di pirimidina nucleotidasi, che può essere dovuta ad un difetto ereditario autosomico recessivo o essere secondaria ad avvelenamento da piombo. La malattia emolitica può manifestarsi in forma acuta nel deficit di glucosio-6-fosfato deidrogenasi o di glutatione reduttasi, mentre negli altri casi assume un andamento cronico; nel deficit di fosfofruttochinasi l’anemia emolitica è generalmente compensata da un aumento dell’eritropoiesi.
Le anemie emolitiche conseguenti a difetti intrinseci della membrana eritrocitaria sono generalmente dovute ad anomalie nelle proteine del citoscheletro (α- e β-spectrina, proteina 4.1, F-actina, anchirina). La sferocitosi ereditaria è una malattia cronica a carattere dominante ma con penetranza variabile, che è caratterizzata dalla presenza in circolo di sferociti con un ridotto rapporto superficie/volume ed associata ad anemia lieve o moderata, modesta spenomegalia ed ittero intermittente. L’ellittocitosi ereditaria è caratterizzata dalla presenza di globuli rossi di forma ellittica con scarsi fenomeni emolitici, mentre la piropoichilocitosi ereditaria dà luogo ad una forma di anemia più grave per la presenza in circolo di poichilociti e di emazie frammentate. L’ellittocitosi e la piropoichilocitosi possono manifestarsi nel medesimo ceppo familiare ed a volte alcuni pazienti con poichilociti ed emolisi durante l’infanzia presentano ellittocitosi in età adulta. Una anemia con microsferocitosi è inoltre osservabile in casi di grave ipofosfatemia ed è dovuta a deplezione dell’ATP intraeritrocitario. Le anemie emolitiche dovute ad emoglobinopatie sono trattate nel Par. 7.2.1.
7.1.3b. Eritrocitosi
Si definiscono eritrocitosi quelle condizioni caratterizzate da un aumento
del numero degli eritrociti circolanti. Le eritrocitosi relative sono
conseguenti a disidratazione o ad altre cause che portano ad una diminuzione
della volemia. Le eritrocitosi secondarie sono dovute ad un aumento appropriato
di eritropoietina
![]() in risposta ad una condizione di ipossia tessutale (elevata
altitudine, sindromi da ipoventilazione, malattie cardio-polmonari,
emoglobinopatie; vedi Par. 7.2.1) o ad un aumento inappropriato di eritropoietina in caso di
neoplasie (ipernefroma, epatoma, mioma uterino, emoangioblastoma, feocromocitoma),
stenosi dell’arteria renale, cisti renali, idronefrosi o trapianto renale. Le
eritricitosi primitive sono dovute ad una crescita incontrollata di cellule
eritroidi con produzione di un eccesso di eritrociti senza motivo apparente (policitemia
vera o morbo di Vaquez, eritremia acuta o malattia di Di Guglielmo).
in risposta ad una condizione di ipossia tessutale (elevata
altitudine, sindromi da ipoventilazione, malattie cardio-polmonari,
emoglobinopatie; vedi Par. 7.2.1) o ad un aumento inappropriato di eritropoietina in caso di
neoplasie (ipernefroma, epatoma, mioma uterino, emoangioblastoma, feocromocitoma),
stenosi dell’arteria renale, cisti renali, idronefrosi o trapianto renale. Le
eritricitosi primitive sono dovute ad una crescita incontrollata di cellule
eritroidi con produzione di un eccesso di eritrociti senza motivo apparente (policitemia
vera o morbo di Vaquez, eritremia acuta o malattia di Di Guglielmo).
7.1.4. Alterazioni della serie bianca
I globuli bianchi sono cellule eterogenee che derivano da una cellula staminale comune che si differenzia in varie linee, ognuna delle quali sviluppa funzioni specifiche. Le malattie dei globuli bianchi possono essere suddivise in malattie non clonali e in malattie clonali. Le malattie non clonali comprendono le alterazioni dei meccanismi di regolazione della produzione di una o più categorie di cellule nel loro complesso. Nelle malattie clonali, generalmente di natura neoplastica, l’alterazione patologica colpisce una singola cellula progenitrice che a sua volta dà origine a un clone cellulare con medesime caratteristiche distintive.
7.1.4a. Alterazioni quantitative non clonali
Si definisce neutropenia una diminuzione del numero assoluto dei neutrofili al di sotto di 2.000 cellule/μL. Si parla di neutropenia lieve se la concentrazione di neutrofili è compresa tra 1.000 cellule/μL e 2.000 cellule/μL, di neutropenia moderata se la concentrazione è compresa tra 500 cellule/μL e 1.000 cellule/μL, di neutropenia grave o di agranulocitosi se la concentrazione è inferiore a 500 cellule/μL. Quest’ultima condizione è tuttavia di raro riscontro in forma pura in quanto molti dei fattori che sopprimono l’attività mielopoietica tendono a deprimere anche la produzione di globuli rossi e delle piastrine.
In alcuni soggetti (fino al 30% dei casi in alcune popolazioni di razza nera)
si può avere una condizione di "neutropenia costituzionale" quando la
concentrazione di neutrofili è normalmente inferiore a 2.000 cellule/μL.
Altre cause di neutropenia sono un deficit di produzione, una eccessiva
distruzione o una anomala distribuzione delle cellule. Il deficit di produzione
può essere congenito (neutropenia familiare benigna, neutropenia ciclica,
agranulocitosi genetica infantile) o acquisito in seguito a carenza di vitamina
B12, acido folico o rame, somministrazione di farmaci o sostanze
citotossiche (chemioterapici, immunosoppressori), infezioni (tifo, epatite,
mononucleosi infettiva, tubercolosi), leucemia, anemia aplastica. L’eccessiva
distruzione delle cellule può essere mediata da reazioni immunitarie (neutropenia
autoimmune, reazioni di citotossicità conseguenti a trasfusioni di sangue,
granulocitopenia alloimmune del neonato, presenza di anticorpi antifarmaci)
oppure non mediata da reazioni immunitarie ed associata a spenomegalia
(ipertensione portale, tesaurismosi, linfomi, artrite reumatoide
![]() ), a malattie
della microcircolazione polmonare o conseguente all’uso di apparati per la
circolazione extracorporea (macchine cuore-polmone, macchine per dialisi
renale). Una anomala distribuzione dei neutrofili può essere conseguente a
splenomegalia o ad un aumento della quota marginata costituita da cellule che
rimangono strettamente aderenti alla parete dei vasi.
), a malattie
della microcircolazione polmonare o conseguente all’uso di apparati per la
circolazione extracorporea (macchine cuore-polmone, macchine per dialisi
renale). Una anomala distribuzione dei neutrofili può essere conseguente a
splenomegalia o ad un aumento della quota marginata costituita da cellule che
rimangono strettamente aderenti alla parete dei vasi.
Si definisce linfocitopenia una diminuzione del numero assoluto di linfociti al di sotto di 1.000 cellule/μL nell’adulto e di 2.500 cellule/μL nel bambino. La linfocitopenia può essere associata a sindromi da immunodeficienza (immunodeficienza umana acquisita da HIV, difetti congeniti dell’immunità cellulo-mediata, terapia immunosoppressiva; vedi Par. 8.6.2a), malattie gravi debilitanti (insufficienza cardiaca congestizia, insufficienza renale, tubercolosi in fase avanzata), difetti della circolazione linfatica (linfangectasia intestinale, disordini della mucosa intestinale, drenaggio del dotto toracico) od aumentati livelli ematici di adrenalina o corticosteroidi (iperattività delle ghiandole surrenali, tumori secernenti ACTH, somministrazione terapeutica di steroidi; vedi Par. 10.5.1d).
Si definisce leucocitosi un aumento del numero dei globuli bianchi oltre
10.000 cellule/μL. Quando questa condizione interessa
una o più categorie di cellule, essa viene identificata con i nomi
specifici di neutrofilia (> 8.000 neutrofili/μL),
eosinofilia (> 450 eosinofili/μL), basofilia (> 50
basofili/μL), linfocitosi (> 4.000 linfociti/μL)
e monocitosi (> 800 monociti/μL). Una leucocitosi può assumere a
volte proporzioni molto rilevanti con immissione in circolo di un gran numero di
elementi maturi ed immaturi. Poiché il quadro periferico è simile a quello
osservabile nella leucemia cronica, questo fenomeno è stato definito "reazione leucemoide", malgrado non si tratti di una alterazione primitiva del midollo, ma
piuttosto di una reazione secondaria a stimoli di varia natura. Le reazioni
leucemoidi più frequenti sono di tipo mieloide
![]() e possono
presentarsi nella fase di recupero da una agranulocitosi o accompagnarsi a
tumori maligni, gravi infezioni piogene o tubercolari, avvelenamenti da metalli
pesanti, gravi disturbi metabolici renali o
epatici, chetoacidosi diabetica, crisi falcemiche (vedi Par.
7.2.1a). Reazioni leucemoidi di tipo linfoide possono
comparire in corso di tubercolosi, pertosse o mononucleosi infettiva. Reazioni
leucemoidi monocitiche sono state descritte in corso di tubercolosi.
e possono
presentarsi nella fase di recupero da una agranulocitosi o accompagnarsi a
tumori maligni, gravi infezioni piogene o tubercolari, avvelenamenti da metalli
pesanti, gravi disturbi metabolici renali o
epatici, chetoacidosi diabetica, crisi falcemiche (vedi Par.
7.2.1a). Reazioni leucemoidi di tipo linfoide possono
comparire in corso di tubercolosi, pertosse o mononucleosi infettiva. Reazioni
leucemoidi monocitiche sono state descritte in corso di tubercolosi.
Condizioni di neutrofilia
possono riscontrasi in casi di stress (prolungato esercizio fisico, stress
emotivo acuto, parto, esposizione a temperature estreme, emorragie acute,
sindromi emolitiche; vedi Par. 7.1.3a), malattie infettive (batteriche, virali, da rickettsie, da
funghi), malattie infiammatorie (febbre reumatica acuta, artrite reumatoide,
gotta acuta, vasculiti, miositi, reazioni da ipersensibilità a farmaci), necrosi
tissutali (ischemie, ustioni, carcinomi e sarcomi), disturbi metabolici (uremia,
chetoacidosi diabetica, eclampsia, crisi tireotossica), somministrazione di
farmaci o veleni. Condizioni di eosinofilia possono riscontrarsi nel corso di
malattie allergiche (asma, febbre da fieno, reazioni a farmaci, vasculite
allergica, malattia da siero), infezioni parassitarie (trichinosi, echinococcosi,
anchilostomiasi, schistosomiasi, amebiasi), malattie della pelle (psoriasi,
penfigo, dermatite erpetiforme), malattie neoplastiche (linfoma di Hodgkin
![]() ,
metastasi diffuse o necrosi di tumori solidi) o in associazione a infiltrazione
polmonare, disturbi cardiovascolari, malattie del collageno associate a vasculite, colite ulcerosa, ipocorticosurrenalisno, sindrome mialgica da
L-triptofano
,
metastasi diffuse o necrosi di tumori solidi) o in associazione a infiltrazione
polmonare, disturbi cardiovascolari, malattie del collageno associate a vasculite, colite ulcerosa, ipocorticosurrenalisno, sindrome mialgica da
L-triptofano
![]() . La basofilia può
essere presente nelle mastocitosi, nelle malattie mieloproliferative e negli
stati di ipersensibilità cronica in assenza dello specifico allergene
. La basofilia può
essere presente nelle mastocitosi, nelle malattie mieloproliferative e negli
stati di ipersensibilità cronica in assenza dello specifico allergene
![]() . Condizioni di linfocitosi
possono riscontrarsi nel corso di malattie infettive (batteriche, virali, da toxoplasma), malattie infiammatorie croniche (colite ulcerosa, malattia da
siero, porpora trombocitopenica idiopatica), alterazioni metaboliche (iposurrenalismo,
ipertiroidismo). Un quadro di monocitosi
si può trovare in caso di malattie infettive (tubercolosi, endocardite batterica subacuta, epatite, malattie da rickettsie, sifilide), malattie granulomatose (sarcoidosi,
colite ulcerosa, enterite regionale), malattie del collageno associate a
vasculopatie (lupus, artrite reumatoide, poliarterite), tumori.
. Condizioni di linfocitosi
possono riscontrarsi nel corso di malattie infettive (batteriche, virali, da toxoplasma), malattie infiammatorie croniche (colite ulcerosa, malattia da
siero, porpora trombocitopenica idiopatica), alterazioni metaboliche (iposurrenalismo,
ipertiroidismo). Un quadro di monocitosi
si può trovare in caso di malattie infettive (tubercolosi, endocardite batterica subacuta, epatite, malattie da rickettsie, sifilide), malattie granulomatose (sarcoidosi,
colite ulcerosa, enterite regionale), malattie del collageno associate a
vasculopatie (lupus, artrite reumatoide, poliarterite), tumori.
7.1.4b. Malattie clonali neoplastiche
Le malattie neoplastiche riguardanti i globuli bianchi sono definite
leucemie. Queste sono state originalmente distinte in acute e croniche in base
alla spettanza di vita; le leucemie acute sono formate principalmente da cellule
immature (usualmente da blasti), mentre le leucemie croniche sono composte da
cellule più mature. Un terzo gruppo, meno aggressivo rispetto ai precedenti, è
costituito dalle sindromi mielodisplastiche
![]() . Sia le
leucemia acute che le croniche sono state suddivise ulteriormente in forme
mieloidi e forme linfoidi (a loro volta distinte in B e T) in base alle
caratteristiche morfologiche ed immunologiche. Le neoplasie originantesi dal
sistema linforeticolare sono state classificate come linfomi, che sono stati
distinti, a loro volta, in due forme principali: il morbo di Hodgkin
. Sia le
leucemia acute che le croniche sono state suddivise ulteriormente in forme
mieloidi e forme linfoidi (a loro volta distinte in B e T) in base alle
caratteristiche morfologiche ed immunologiche. Le neoplasie originantesi dal
sistema linforeticolare sono state classificate come linfomi, che sono stati
distinti, a loro volta, in due forme principali: il morbo di Hodgkin
![]() (suddiviso in
quattro sottotipi istopatologici) e il linfomi non-Hodgkin (comprendenti un
gruppo più eterogeneo di malattie).
(suddiviso in
quattro sottotipi istopatologici) e il linfomi non-Hodgkin (comprendenti un
gruppo più eterogeneo di malattie).
La moderna classificazione delle neoplasie dei tessuti emopoietici si basa sull’analisi morfologica degli elementi figurati del sangue e del midollo osseo, sugli studi di citogenetica, immunogenetica e biologia molecolare (ad esempio attraverso tecniche di ibridazione in situ, vedi Par. 14.6.6d), nonché sull’esame del quadro clinico e dei trial terapeutici. L’attuale nomenclatura è stata proposta nel 2000 dalla Organizzazione Mondiale della Sanità (WHO, World Health Organization) ed è una rielaborazione della precedente classificazione ideata negli anni 1976-1994 dal gruppo FAB (French-American-British Group), della classificazione proposta dal gruppo operativo MIC (Morphologic, Immunologic, Cytogenetic Cooperative Study Group) e della classificazione denominata con l’acronimo REAL (Revised European-American Lymphoma Classification). La classificazione dei principale gruppi di neoplasie è riportata in Tab. 7.IIII. Per una completa classificazione dei diversi sottogruppi di leucemie, si rimanda il lettore a trattati specialistici di ematologia.
Tab. 7.III. Classificazione delle neoplasie dei tessuti emopoietici secondo il WHO
| Neoplasie mieloidi | |
| malattie mieloproliferative | |
| malattie dismieloproliferative | |
| malattie mielodisplastiche | |
| leucemie acute mieloidi | |
| leucemie acute mieloidi non-classificate | |
| leucemie acute bifenotipiche | |
| Neoplasie delle cellule B | |
| neoplasie dei precursori delle cellule B | |
| neoplasie delle cellule B mature (periferiche) | |
| Neoplasie delle cellule T | |
| neoplasie dei precursori delle cellule T | |
| neoplasie delle cellule T mature (periferiche) | |
| Linfoma di Hodgkin | |
| Malattie delle mast-cellule | |
| Neoplasie delle cellule istiocitiche e dendritiche | |
| sarcoma istiocitico | |
| neoplasie delle cellule dendritiche | |
I grafici di distribuzione della serie bianca, ottenuti mediante citometria a flusso, permettono una iniziale grossolana classificazione delle leucemie e possono essere d’aiuto per indirizzare successivi studi più approfonditi attraverso l’uso di anticorpi monoclonali. Le leucemie mieloidi possono essere differenziate dalle leucemie linfoidi misurando, su un apposito canale, la perossidasi presente nelle cellule (vedi Par. 7.1.2b) in quanto i linfociti di norma sono privi di questo enzima. I citogrammi ottenibili con tale tecnica sono molto eterogenei e riflettono i differenti livelli di differenziazione delle cellule mieloidi per quanto riguarda lo sviluppo dell’attività perossidasica (Fig. 7.6).
|
Il citogramma ottenuto misurando il volume dei leucociti in funzione dell’attività perossidasica permette di distinguere 7 diversi quadri (da P0 a P6). Le aree indicate nello schema corrispondono a successivi livelli di differenziazione delle cellule leucemiche. |
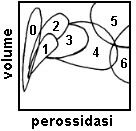 |
P0: Assenza di attività perossidasica con mancata differenziazione mieloide. Il quadro è indicativo di leucemia linfoide acuta o cronica, linfoma non-Hodgkin, linfocitosi virale atipica o leucemia mieloide acuta (sottotipi M0, M5a, M6, M7). |
 |
|
P1: La distribuzione delle cellule nel citogramma si allarga leggermente verso l’alto, indicando la presenza di un basso numero di elementi con attività perossidasica ed iniziale o parziale differenziazione mieloide. Il quadro è indicativo di leucemia mieloide acuta (sottotipi M1, M2, M5a, M5b). |
 |
P2: Un gruppo più o meno omogeneo di cellule invade lo spazio destinato ai neutrofili nel citogramma, superando l’area dei monociti, Il quadro è indicativo di leucemia mieloide acuta (sottotipi M1, M2, M4, M5a, M5b) e deficit parziale di mieloperossidasi. |
 |
|
P3: L’attività perossidasica è da moderata a forte. La dimensione delle cellule è omogenea. Il quadro è indicativo di leucemia mieloide acuta (sottotipi M2, M4). |
 |
P4: Il citogramma mostra una forte ed eterogenea attività perossidasica ed è indicativo di leucemia mieloide cronica o acuta (sottotipi M2, M4). |
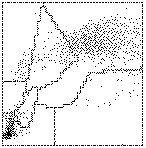 |
|
P5: Il citogramma mostra una forte attività perossidasica in cellule di grosse dimensioni. Il quadro è indicativo di sindrome mielodisplastica, leucemia mieloide cronica o acuta (sottotipo M3v). |
 |
P6: Il citogramma mostra cellule con attività perossidasica estremamente elevata ed è indicativo di leucemie mieloide acuta (sottotipo M3). |
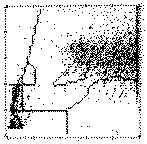 |
Fig. 7. 6. Distribuzione degli elementi figurati nel citogramma ottenuto sul canale della perossidasi. Le leucemie acute mieloidi sono state distinte in base alla classificazione proposta dal gruppo FAB, che comprende 8 sottotipi definiti con una lettera M affiancata da un numero tra 0 e 7.
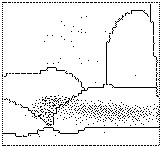
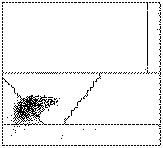
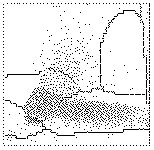
Il citogramma ricavato sul canale dei basofili (vedi Par. 7.1.2b) fornisce un indice di densità nucleare e un indice di volume che sono utilizzabili per distinguere le leucemie acute dalle leucemie croniche e dalle linfocitosi virali atipiche. Nel caso delle leucemie acute, gli elementi cellulari sono caratterizzati da nuclei immaturi che si posizionano nella porzione sinistra del tracciato, mentre nelle leucemie croniche la differenziazione cellulare è più avanzata e ciò comporta una distribuzione degli elementi figurati simile a quella ottenibile da un soggetto sano (Fig. 7.7).
 |
 |
 |
||
|
D0: La distribuzione delle cellule mononucleate è normale. In un paziente leucemico, il quadro è indicativo di una forma cronica (leucemia mieloide o linfoide cronica). |
D1: La distribuzione delle cellule è spostata nella zona inferiore sinistra del citogramma per una probabile presenza di elementi con cromatina immatura. Il quadro è indicativo di una leucemia acuta mieloide o linfoide. Quando sono presenti dei blasti, la distribuzione delle cellule assume questo aspetto. |
D2: Lo spostamento della distribuzione delle cellule nel citogramma verso l’alto è indicativo di una linfocitosi virale atipica e, in particolare, di una mononucleosi infettiva. Sono presenti cellule di grosse dimensioni con cromatina eterogenea, mentre i blasti sono di norma assenti. |
||
Fig. 7.7. Distribuzione degli elementi figurati nel citogramma ottenuto sul canale dei basofili. Il citogramma ottenuto sul canale dei basofili permette di classificare tre quadri (da D0 a D2) caratterizzati da diversa densità della cromatina nucleare e quindi da un diverso grado di maturazione.
7.1.5. Alterazioni quantitative delle piastrine
Il numero delle piastrine nel sangue può diminuire oppure aumentare nel corso di processi patologici. Nel primo caso si parla di trombocitopenia, mentre nel secondo caso si parla di trombocitosi o di trombocitemia.
7.1.5a. Trombocitopenia
La trombocitopenia può essere dovuta a diminuita produzione midollare, ad aumentato sequestro periferico o ad accelerata distruzione delle piastrine. Un basso valore di conteggio delle piastrine può anche essere dovuto ad artefatti (pseudotrombocitopenia) in quanto le piastrine possono aggregare in ammassi dopo il prelievo, soprattutto se il sangue è stato reso incoagulabile con EDTA (vedi Par. 7.1.6).
In genere, le condizioni morbose che alterano i processi maturativi delle
cellule midollari causano, tra le altre manifestazioni ematologiche, anche
trombocitopenia. Per tale motivo la trombocitopenia si accompagna frequentemente
ad anemia (vedi Par. 7.1.3a) e leucopenia (vedi
Par. 7.1.4a). La trombocitopenia può dipendere in qualche
raro caso da un deficit isolato di megacariociti per un difetto primario
![]() , per un
difetto secondario alla presenza di una sindrome mielodisplastica o per la
distruzione autoimmune dei precursori piastrinici in pazienti con una malattia
del collageno. Sostanze che deprimono selettivamente la produzione di piastrine
sono la clorotiazide
, per un
difetto secondario alla presenza di una sindrome mielodisplastica o per la
distruzione autoimmune dei precursori piastrinici in pazienti con una malattia
del collageno. Sostanze che deprimono selettivamente la produzione di piastrine
sono la clorotiazide
![]() e l’alcool
etilico
e l’alcool
etilico
![]() .
.
Un aumento massivo di volume della milza causa una diminuzione del numero di piastrine, che vengono rimosse dal circolo, e riduce il loro tempo di sopravvivenza. Cause frequenti di spenomegalia sono le epatopatie, l’ipertensione portale e i linfomi. Una drammatica diminuzione di piastrine in circolo può inoltre comparire in tutte quelle situazioni in cui vi sia un esteso danno endoteliale (vasculiti, sepsi).
Una accelerata distruzione delle piastrine può essere su base immunitaria o
dovuta ad altri fattori. Le piastrine possono essere distrutte per la presenza
di alloanticorpi (piastrinopenia neonatale alloimmune, porpora
post-trasfusionale), di autoanticorpi (trombocitemia autoimmune idiopatica o
secondaria ad altre malattie
![]() ) o di
anticorpi diretti contro farmaci che si adsorbono sulle piastrine stesse (chinidina,
digitossina, tiazidici, eparina). Distruzioni piastriniche non direttamente
riconducibili a cause autoimmuni si riscontrano nella coagulazione intravasale disseminata (vedi Par. 7.3.3c), nella porpora trombotica trombocitopenica
) o di
anticorpi diretti contro farmaci che si adsorbono sulle piastrine stesse (chinidina,
digitossina, tiazidici, eparina). Distruzioni piastriniche non direttamente
riconducibili a cause autoimmuni si riscontrano nella coagulazione intravasale disseminata (vedi Par. 7.3.3c), nella porpora trombotica trombocitopenica
![]() e nella
sindrome emolitico-uremica
e nella
sindrome emolitico-uremica
![]() .
.
7.1.5b. Trombocitosi e trombocitemia
Con il termine "trombocitosi" si indica un aumento del numero delle piastrine circolanti conseguente ad una risposta secondaria o reattiva. Con il termine "trombocitemia" si indica una iperproduzione incontrollata di piastrine, come quella che si verifica in corso di sindromi mieloproliferative. La trombocitosi è un reperto comune in molti processi morbosi in quanto accompagna spesso processi infiammatori, infezioni, tumori maligni e la fase di recupero dopo una emorragia acuta o può essere conseguente alla mobilitazione dei depositi piastrinici (in seguito a splenectomia o a somministrazione di adrenalina). Sindromi mieloproliferative, causa di trombocitemia, sono la trombocitemia essenziale, la policitemia vera, la mielofibromatosi idiopatica con metaplasia mieloide e la leucemia mieloide cronica.
7.1.6. Preparazione del campione ed intervalli di riferimento
Il sangue utilizzato per l’esame emocromocitometrico deve essere reso incoagulabile. A tale scopo può essere aggiunto dell’ossalato d’ammonio o di potassio, ma è preferibile una combinazione di entrambi i sali per ridurre l’alterazione delle cellule del sangue ed in particolare dei globuli rossi. L’acido etilendiaminico tetracetico (EDTA), sotto forma di sale sodico o potassico, è preferibile agli ossalati in quanto preserva maggiormente gli elementi figurati.
Per allestire gli strisci di sangue, è più opportuno non usare il sangue trattato con anticoagulanti, anche se è possibile eseguire eventualmente uno striscio con il sangue in ossalato o in EDTA, purché questo venga effettuato non oltre 5 minuti dal prelievo di sangue. Appare assolutamente controindicato l’uso del sangue in eparina perchè questa sostanza interferisce con la colorazione delle cellule.
I valori di riferimento dei vari parametri ematici sono riportati in Tab. 7.1. Un esempio di profilo ematologico normale è riportato in Fig. 7.8.
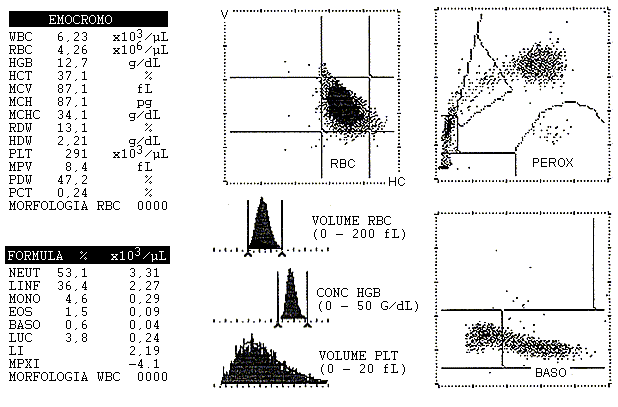
Fig. 7.8. Esempio di un profilo ematologico normale. Gli eritrociti e le piastrine sono stati esaminati mediante un rivelatore a diffusione luminosa (vedi Par. 7.1.2a). I globuli bianchi sono stati esaminati sul canale della perossidasi e dei basofili (vedi Par. 7.1.2b). L’indice di lobularità (LI) è un numero puro ed ha un intervallo di riferimento compreso fra 1,9 e 3,0. L’indice di attività perossidasica media (MPXI) è espresso come variazione percentuale rispetto ad un campione standard ed ha un intervallo di riferimento compreso fra -10 e +10. La morfologia delle cellule è valutata dal computer in linea con l’apparecchio in base a dei parametri standard. Il significato delle altre sigle riportate in tabella è indicato nel testo (vedi Par. 7.1).
7.2. EMOGLOBINA
Tutte le forme normali di emoglobina hanno una struttura tetramerica costituita da due coppie disuguali di catene polipeptidiche, che vengono generalmente siglate con lettere greche. Nell’adulto, circa il 97% dell’emoglobina è costituito dalla forma HbA (α2β2), circa il 3% dalla forma HbA2 (α2δ2), mentre la forma HbF (α2γ2) è presente solo in tracce (costituisce circa il 0,5% dell’emoglobina totale). Altre varianti dell’emoglobina derivano da modificazioni della catena aminoacidica successive alla sintesi: la glicazione dell’HbA porta alla formazione delle varianti HbA1 (vedi Par. 2.2), mentre l’acetilazione della HbF porta alla HbF1.
Le forme di emoglobina sintetizzate nel corso dello sviluppo embrionale e fetale sono diverse. Nelle prime fasi di sviluppo embrionale vengono prodotte le emoglobine Gower 1 (ζ2ε2), Gower 2 (α2ε2) e Portland (ζ2γ2). Successivamente, verso l’ottava settimana di gestazione, inizia la sintesi epatica di HbF e di piccole quantità (< 10%) di HbA. Dopo la diciottesima settimana il fegato è progressivamente sostituito dal midollo osseo nella produzione dei globuli rossi e la sintesi di HbF diminuisce, mentre quella dell’HbA aumenta. L’aumento percentuale di HbA continua dopo la nascita e alla fine del primo anno di vita il livello di HbF è inferiore al 2% rispetto il totale.
In generale, le catene costituenti le emoglobine possono essere classificate in catene tipo-α (α e ζ, ciascuna di 141 aminoacidi) e in catene tipo-β (β, γ, δ e ε, ciascuna di 146 aminoacidi). Le catene tipo-α sono codificate a livello della banda 16p13.3 all’estremità del cromosoma 16. I geni delle catene tipo-β si trovano sul cromosoma 11 a livello di un cluster nella parte distale della banda 11p14.
7.2.1. Emoglobinopatie
Le alterazioni riguardanti l’emoglobina possono interessare (a) la struttura aminoacidica delle catene o (b) la velocità con la quale sono sintetizzate le varie subunità. Nel primo caso viene prodotta una emoglobina con proprietà anomale. Nel secondo caso si ha uno sbilanciamento nella sintesi delle differenti catene dell’emoglobina e la malattia è definita come sindrome talassemica. Sebbene sia utile dal punto di vista concettuale, questa classificazione non è del tutto soddisfacente in quanto alcune varianti strutturali possono anche essere sintetizzate più lentamente o dare luogo a subunità fortemente instabili con un quadro clinico di tipo talassemico. Inoltre, in diverse popolazioni vi è un’alta incidenza sia di varianti strutturali dell’emoglobina sia di forme di talassemia. Non è perciò raro che un individuo erediti entrambi i difetti con manifestazioni cliniche complesse.
7.2.1a. Varianti strutturali
Sono state descritte finora circa 750 varianti strutturali. Nella maggioranza
dei casi l’alterazione nella struttura riguarda un singolo aminoacido, in altri
casi la catena polipeptidica della globina è più corta o più lunga del normale,
in altri casi ancora la globina alterata è il prodotto di una fusione fra una
parte della catena β e una parte della catena δ o una parte della catena β
e una parte della catena γ. L’alterazione strutturale
può causare un’anemia emolitica
![]() o una metaemoglobinemia congenita, oppure
aumentare o diminuire l’affinità dell’emoglobina per l’ossigeno
o una metaemoglobinemia congenita, oppure
aumentare o diminuire l’affinità dell’emoglobina per l’ossigeno
![]() . La malattia si
manifesta generalmente negli omozigoti o nell’eterozigosi composita (compound
heterozygosis) per la presenza contemporanea di due varianti patologiche nel
medesimo soggetto. Le varianti che sono presenti nella popolazione con maggiore
frequenza interessano la catena β (HbS, HbC, HbE). Le
varianti HbS e HbC sono presenti in popolazioni di derivazione africana, mentre
la variante HbE è presente in particolare nel sud-est asiatico nella fascia che
si estende dalla Tailandia al Burna.
. La malattia si
manifesta generalmente negli omozigoti o nell’eterozigosi composita (compound
heterozygosis) per la presenza contemporanea di due varianti patologiche nel
medesimo soggetto. Le varianti che sono presenti nella popolazione con maggiore
frequenza interessano la catena β (HbS, HbC, HbE). Le
varianti HbS e HbC sono presenti in popolazioni di derivazione africana, mentre
la variante HbE è presente in particolare nel sud-est asiatico nella fascia che
si estende dalla Tailandia al Burna.
L’emoglobina HbS differisce dalla HbA per una valina al posto dell’acido glutammico in posizione 6 sulla catena β e ciò determina una diminuzione della carica elettrica della proteina, che migra a pH alcalino verso l’anodo più lentamente della HbA. L’emoglobina HbS, quando è deossigenata, ha una solubilità abnormemente bassa e dà luogo un precipitato fibroso che deforma i globuli rossi e conferisce loro un aspetto a falce. I globuli rossi a falce vengono intrappolati nei piccoli vasi sanguigni, compromettendo la circolazione specialmente a livello delle ossa e dei reni. Le cellule falciformi sono inoltre più fragili di quelle normali e vanno incontro ad emolisi determinando un grave forma di anemia (anemia falciforme o drepanocitica).
L’emoglobina HbC differisce dalla HbA per una lisina al posto dell’acido glutammico in posizione 6 sulla catena β. Il difetto strutturale causa un’anemia emolitica accompagnata a splenomegalia. L’eterozigosi composita Hb S-C dà luogo ad una forma più lieve di anemia; i sintomi sono simili a quelli dell’anemia falciforme, ma meno gravi. L’eterozigosi composita HbC-β° talassemia ha manifestazioni cliniche simili a quelle presentate dagli omozigoti per l’emoglobina HbC.
L’emoglobina HbE differisce dalla HbA per una lisina al posto dell’acido glutammico in posizione 26 sulla catena β. Questa variante è sintetizzata con minore efficienza e dà luogo ad una forma lieve di β talassemia (vedi Par. 7.2.1b). Gli eterozigoti non presentano segni clinici ed hanno un normale livello di emoglobina, sebbene i globuli rossi siano livemente microcitici ed ipocromici. Gli omozigoti presentano solo una leggera anemia e degli indici ematologici simili a quelli presenti negli eterozigoti per la β talassemia. Soggetti con eterozigosi composita HbE-β° talassemia hanno il 50-70% dell’emoglobina come HbF e il rimanente come HbE. L’eterozigosi composita HbE-β+ talassemia comporta disturbi meno gravi e una quantità variabile di HbA nei globuli rossi.
7.2.1b. Sindromi talassemiche
Le talassemie costituiscono un gruppo eterogeneo di disordini caratterizzati dall’assenza o da una ridotta sintesi di una o più globine, una situazione che porta, all’interno della cellula, ad un rapporto sbilanciato fra le varie subunità dell’emoglobina. I più comuni e più importanti tipi di talassemia sono l’α, la β e la δβ talassemia. Ciascun tipo comprende quelle forme che sono caratterizzate dalla completa assenza di sintesi di una particolare globina, che sono denominate rispettivamente α°, β° e (δβ)° talassemie, e quelle forme nelle quali la catena è ancora sintetizzata, ma ad una velocità ridotta, che sono denominate rispettivamente α+, β+ e (δβ)+ talassemie. La sindrome da persistenza ereditaria di emoglobina fetale può essere considerata una forma particolarmente lieve di β o δβ talassemia, dove il difetto di sintesi della catena β è completamente compensato da una persistente sintesi della catena γ oltre il periodo neonatale.
In alcune popolazioni, possono essere presenti diversi tipi di talassemia e questi possono coesistere, a loro volta, con altri difetti congeniti dovuti ad alterazioni strutturali dell’emoglobina. Ciò rende possibile che uno stesso individuo erediti i geni relativi a più di un tipo di talassemia e/o sia portatore di alterazioni sia di tipo talassemico che di tipo strutturale. L’interazione fra i diversi tipi di emoglobinopatie porta a manifestazioni cliniche estremamente diversificate, che nel complesso sono indicate con il nome di sindromi talassemiche.
Nei soggetti omozigoti per la β talassemia, la sintesi della catena β è assente (β° talassemia) o fortemente ridotta (β+ talassemia). Le catene α, che non possono essere utilizzate per formare un tetramero di emoglobina, tendono a precipitare nelle cellule che vengono così distrutte. La morte delle cellule eritroidi in corso di maturazione nel midollo osseo dà luogo ad una eritropoiesi inefficace, mentre la distruzione dei globuli rossi maturi, che sono ipocromici e microcitici a causa della ridotta sintesi di emoglobina, comporta una emolisi intravasale. Poiché il difetto riguarda le catene β, la sintesi di HbF ed HbA2 non è compromessa. Lo sviluppo fetale è normale e la malattia si manifesta quando in epoca neonatale si passa dalla sintesi di catene γ alla sintesi di catene β. I soggetti eterozigoti per la β talassemia hanno un minore sbilanciamento nella sintesi delle varie catene di emoglobina, così che le catene in eccesso possono essere distrutte dagli enzimi proteolitici presenti nelle cellule eritroidi riducendo di molto l’eritropoiesi inefficace.
Nei soggetti affetti da α talassemia, è la sintesi delle catene α ad essere assente (α° talassemia) o fortemente ridotta (α+ talassemia). Poiché le catene α sono presenti sia nell’emoglobina fetale che in quella dell’adulto, il difetto si manifesta già prima della nascita. La carenza di catene α porta nel feto ad relativa sopraproduzione di catene γ che polimerizzano formando un omotetramero γ4 (emoglobina di Bart), nell’adulto ad una relativa sopraproduzione di catene β che polimerizzano formando un omotetramero β4 (HbH). Sia l’emoglobina di Bart che l’HbH hanno un’altissima affinità e curve di dissociazione per l’ossigeno di tipo quasi iperbolico e quindi non sono capaci di cedere ossigeno ai tessuti in condizioni fisiologiche. La grave carenza di ossigenazione dei tessuti causa uno stato idropico nel feto (per aumento della permeabilità capillare conseguente all’ipossia) ed una enorme ipertrofia placentare. Entrambi gli omotetrameri γ4 e β4 sono solubili nelle cellule eritroidi in corso di maturazione e per questo motivo l’α talassemia non è caratterizzata da una grave forma di eritropoiesi inefficace. Tuttavia, mentre l’emoglobina di Bart è stabile, i tretrameri β4 tendono a precipitare nei globuli rossi invecchiati e, per questo motivo, l’anemia presente nelle forme più gravi di α talassemia dell’adulto è in gran parte dovuta ad una distruzione prematura delle emazie a livello splenico. Inoltre, a causa del difetto di sintesi dell’emoglobina, i globuli rossi sono generalmente ipocromici e microcitici.
7.2.2. Metodi di determinazione
L’emoglobina viene determinata sul sangue intero opportunamente diluito in una soluzione litica ipotonica. La lisi dei globuli rossi è facilitata dall’aggiunta di un detergente non-ionico, utile anche per ridurre l’eventuale torbidità della soluzione dovuta alla presenza di lipoproteine e di membrane dei globuli rossi. La concentrazione viene calcolata dall’assorbimento ottico della soluzione di emoglobina o di un suo derivato.
Il metodo più comunemente usato consiste nel convertire l’emoglobina in
cianometaemoglobina mediante aggiunta di cianuro di potassio e ferricianuro di
potassio e nel determinare l’assorbimento della soluzione a 540 nm. La reazione
è accelerata acidificando la soluzione con l’aggiunta di fosfato monopotassico.
L’emoglobina, la metaemoglobina e la carbossiemoglobina sono tutte convertite in
cianometaemoglobina. La sulfoemoglobina non subisce la trasformazione e ciò può
portare ad una sottostima della concentrazione dell’emoglobina totale quando la
sulfoemoglobina è particolarmente concentrata nel campione biologico
![]() . La
presenza di carbossiemoglobina può invece portare ad una sovrastima
dell’emoglobina totale in quanto la carbossiemoglobina ha un coefficiente di
assorbimento a 540 nm maggiore di quello della cianometaemoglobina e viene
convertita in quest’ultimo composto piuttosto lentamente
. La
presenza di carbossiemoglobina può invece portare ad una sovrastima
dell’emoglobina totale in quanto la carbossiemoglobina ha un coefficiente di
assorbimento a 540 nm maggiore di quello della cianometaemoglobina e viene
convertita in quest’ultimo composto piuttosto lentamente
![]() . Anche la torbidità del campione può dare luogo a
risultati erronei. La torbidità dei campioni iperlipemici può essere
eliminata estraendo i grassi in etere dietilico. Se il campione è torbido per la
presenza di paraproteine, può essere chiarificato mediante l’aggiunta di carbonato di
potassio o di una soluzione di ammoniaca diluita. Se la torbidità è dovuta ai
globuli bianchi, questi possono essere allontanati mediante centrifugazione o
filtrazione. Se i globuli rossi sono lisati solo parzialmente (le cellule
contenenti emoglobina S o C si lisano con maggiore difficoltà), si può diluire il
campione in un eguale volume di acqua distillata.
. Anche la torbidità del campione può dare luogo a
risultati erronei. La torbidità dei campioni iperlipemici può essere
eliminata estraendo i grassi in etere dietilico. Se il campione è torbido per la
presenza di paraproteine, può essere chiarificato mediante l’aggiunta di carbonato di
potassio o di una soluzione di ammoniaca diluita. Se la torbidità è dovuta ai
globuli bianchi, questi possono essere allontanati mediante centrifugazione o
filtrazione. Se i globuli rossi sono lisati solo parzialmente (le cellule
contenenti emoglobina S o C si lisano con maggiore difficoltà), si può diluire il
campione in un eguale volume di acqua distillata.
Metodi alternativi per il dosaggio dell’emoglobina consistono nell’aggiungere al campione il laurilsolfato di sodio e misurare l’assorbimento della luce a 534 nm oppure il nitrato di sodio e l’azide sodica e misurare l’assorbimento della luce a 570 nm. L’emoglobina può inoltre essere misurata come ematina in condizioni alcaline. Quest’ultimo metodo permette di determinare contemporaneamente la carbossiemoglobina, la sulfoemoglobina e la metaemoglobina, ma è inadatto per determinare l’emoglobina fetale (HbF) o l’emoglobina di Bart (vedi Par. 7.2.1b) che sono particolarmente resistenti alla denaturazione alcalina.
L’emoglobina può essere determinata direttamente senza dover subire modificazioni chimiche misurando l’assorbimento a 548,5 nm. A questa lunghezza d’onda la deossiemoglobina e l’ossiemoglobina hanno il medesimo coefficiente di assorbimento, mentre quello della carbossiemoglobina è appena inferiore. Un altro metodo consiste nell’integrare l’assorbimento del campione tra 500 e 600 nm in quanto l’assorbimento integrale della deossiemoglobina, della ossiemoglobina e della carbossiemoglobina è molto simile in questo intervallo.
7.2.2a. Elettroforesi dell’emoglobina
L’elettroforesi è una delle metodiche più usate per la separazione e l’identificazione delle varie forme di emoglobina. I tamponi più comunemente adoperati sono il veronal (acido dietilbarbiturico) o il Tris (tris-idrossimetil aminometano) a pH 8,6 - 9,5. Solo per ottenere una più precisa identificazione di alcune emoglobine (HbC, HbE, HbS) si ricorre all’impiego di un tampone citrato a pH 6,2. I supporti più usati sono l’acetato di cellulosa, il gel d’agar ed il gel di poliacrilamide.
7.3. EMOCOAGULAZIONE E FIBRINOLISI
La coagulazione del sangue è assicurata da un complesso sistema di proteine plasmatiche che permettono l’arresto del processo emorragico attraverso una cascata di reazioni enzimatiche che portano alla formazione di un gel costituito da un reticolo di fibrina elastico e resistente ai traumi. Questo sistema è in stretto rapporto funzionale con i fattori vascolari che sovrintendono all’emostasi, con le piastrine e con il sistema fibrinolitico.
L’endotelio e lo strato subendoteliale della parete vasale svolgono un ruolo centrale nel mantenimento dell’equilibrio emostatico. Le cellule endoteliali ricoprono tutta la superficie interna dei vasi e sono rivestite da una strato glicoproteico, comune per altro a tutti i tipi di cellule, che rappresenta la struttura chimica responsabile dei fenomeni di contatto di superficie. Le cellule endoteliali sono inoltre ricche di attivatore del plasminogeno, che viene sintetizzato e liberato continuamente in circolo, e di altri fattori che intervengono nella cascata coagulativa. La membrana basale contiene fibre collagene, mucopolisaccaridi, microfibrille, elastina, tutti componenti estremamente importanti per la resistenza meccanica della parete e per l’interazione con le piastrine circolanti. La presenza di fibrocellule muscolari lisce nello strato medio delle arterie spiega l’efficacia della contrazione vascolare nel controllo iniziale del sanguinamento. La riduzione del lume vasale, per contrazione riflessa a livello arteriolare e venoso e per rigonfiamento delle cellule endoteliali a livello capillare, rappresenta la "fase vasale" del processo emostatico. Questa fase, che è di breve durata (30-60 sec), è sostenuta sia da un riflesso asso-assonico o da un riflesso spinale, sia da mediatori chimici, come la serotonina liberata dalle piastrine, il fibrinopeptide B derivato dal fibrinogeno per azione della trombina (vedi Par. 7.3.1f), la callicreina e la bradichinina che si formano per attivazione del fattore XII (vedi Par. 7.3.1g).
La soluzione di continuo dell’endotelio vasale o comunque la perdita della proprietà idrorepellente dell’endotelio determina una serie di reazioni a catena il cui primo evento è rappresentato dall’adesione piastrinica. Le piastrine presentano sulla propria membrana una grande varietà di recettori capaci di interagire con le componenti della matrice extracellulare quando viene lesa la barriera costituita dalle cellule endoteliali. L’adesione delle piastrine è facilitata da un processo di attivazione delle piastrine stesse che perdono l’originaria forma discoidale ed assumono un aspetto ameboide. Le piastrine attivate espongono sulla propria superficie delle proteine, normalmente contenute nei granuli citoplasmatici, che aumentano le proprietà adesive della membrana. L’aggregazione delle piastrine porta alla formazione del trombo bianco, caratteristico della "fase piastrinica" del processo emostatico, che può permettere l’arresto del sanguinamento, ma non è molto stabile né resistente ai traumi. Esponendo sulla superficie i fosfolipidi di membrana, le piastrine contribuiscono alla formazione di importanti complessi intermedi del processo coagulativo durante la "fase plasmatica" del processo emostatico ed offrono nel contempo una superficie per le reazioni enzimatiche che portano alla fibrinogenesi. Non è ancora chiaro se le piastrine attivate possano, a loro volta, attivare direttamente la cascata coagulativa, ma è probabile che le piastrine stimolate dal collageno attivino il fattore XI, innescando la via intrinseca della coagulazione (vedi Par. 7.3.1g).
7.3.1. Emocoagulazione
Il processo che porta alla coagulazione del sangue viene schematicamente distinto in due vie, una estrinseca ed una intrinseca, convergenti in una via comune (Fig. 7.9). La via estrinseca viene attivata dal fattore tessutale (tromboplastina tessutale) che normalmente non si trova nel plasma, ma che comunque è sempre necessario perché possa scatenarsi il processo coagulativo in vivo. Nella via intrinseca intervengono invece delle proteine già presenti normalmente nel plasma, denominate fattori di contatto (fattore XII, fattore XI, precallicreina, chininogeno ad alto peso molecolare), che sono attivate quando il sangue viene in contatto con superfici non ricoperte da endotelio.
Nel suo insieme, la coagulazione del sangue avviene attraverso un complicato processo che richiede spesso la formazione di complessi multifattoriali comprendenti enzimi, fattori plasmatici, fosfolipidi e ioni calcio. In questo processo intervengono componenti che appartengono sia alla via estrinseca che alla via intrinseca, in modo tale che difetti di fattori di una delle due vie non possono essere compensati in vivo dalla normale attivazione dell’altra via.
|
|
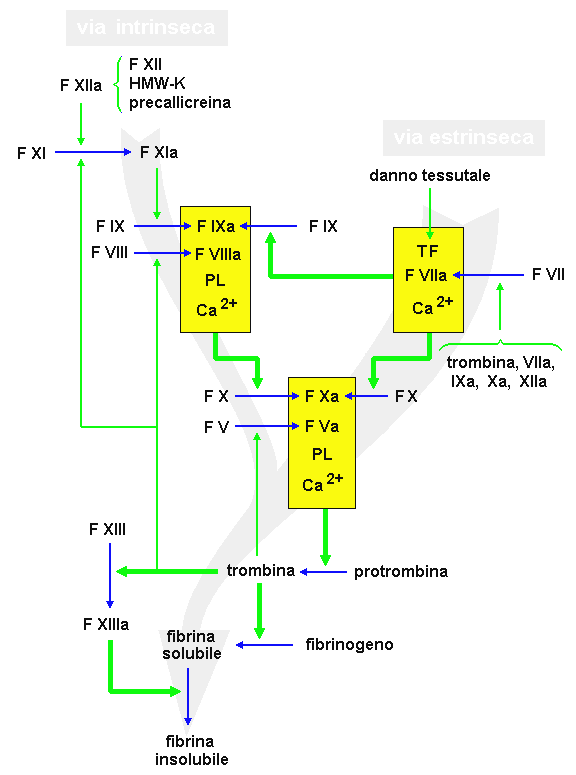
Le proteine che intervengono nella coagulazione del sangue sono sintetizzate in larga misura dagli epatociti, dai megacariociti, dalle cellule endoteliali e, probabilmente, dai macrofagi.
I fattori II, VII, IX e X, la proteina C anticoagulante, la proteina S e la proteina Z sono di origine epatica e dipendono strettamente dalla presenza di vitamina K. Infatti, una volta sintetizzate le catene polipeptidiche, ciascuna di queste proteine viene modificata dall’enzima γ-glutamil carbossilasi, che opera in presenza di vitamina K, anidride carbonica ed ossigeno atmosferico catalizzando l’incorporazione di un secondo gruppo carbossilico in posizione γ sui residui di acido glutammico della catena aminoacidica (Fig. 7.10). Nel corso della reazione, la vitamina K viene prima ossidata a 2,3-epossido e, successivamente, ridotta nuovamente ad idrochinone ad opera di una reduttasi specifica. I residui di acido γ-carbossiglutammico sono utili alle proteine per complessare gli ioni calcio, necessari perché le proteine stesse possano svolgere il proprio compito lungo la cascata coagulativa. Nel corso di avitaminosi K o di terapie che prevedono l’uso di anticoagulanti orali ad attività anti-vitamina K, sono rilasciate dagli epatociti delle proteine prive di acido γ-carbossiglutammico e perciò funzionalmente inattive (PIVKA, proteins induced by vitamin K absence or antagonists).
|
|
Il fibrinogeno è sintetizzato negli epatociti e nei megacariociti, mentre l’antitrombina III è sintetizzata negli epatociti e nelle cellule endoteliali. Il fattore V è sintetizzato esclusivamente negli epatociti, ma è presente anche nelle piastrine in quanto esse lo adsorbono dal plasma sulla loro membrana. Il fattore von Willebrand è sintetizzato dalle cellule endoteliali. Non si hanno sufficienti dati sulla sintesi degli altri fattori, anche se il fegato sembra un organo importante per la sintesi dei fattori XI, XII e XIII. Quest’ultimo fattore è formato anche dai megacariociti, mentre il fattore VIII è probabilmente sintetizzato a livello delle cellule macrofagiche.
7.3.1a. Formazione del complesso costituito da fattore tessutale, fattore VIIa e ioni calcio
La tromboplastina tessutale (fattore tessutale o fattore III) è una fosfoglicoproteina di 44 kDa presente nelle membrane di tutte le cellule, ad eccezione delle piastrine. La proteina non è normalmente esposta sulla superficie della membrana plasmatica, ma lo diventa quando le cellule vengono modificate o quando le cellule normalmente non in contatto con il sangue (come i fibroblasti o le cellule muscolari) sono esposte ad esso. La tromboplastina tessutale è prodotta anche dalle cellule endoteliali stimolate dalla trombina, dall’interleuchina-1 o dalle endotossine.
La tromboplastina tessutale funge da recettore per il fattore VII (proconvertina), una glicoproteina di 50 kDa costituita da una unica catena di aminoacidi, che diventa enzimaticamente attiva quando viene esposta a proteasi presenti nel plasma (trombina, fattori VIIa, IXa, Xa, XIIa) capaci di scindere la catena in due frammenti che rimangono legati da un ponte disulfuro (fattore VIIa). Si viene così a formare un complesso costituito dalla tromboplastina, dal fattore VIIa e dagli ioni calcio, capace di trasformare i fattori IX e X nei corrispondenti fattori attivi.
7.3.1b. Formazione del complesso costituito da fattore IXa, fattore VIIIa, fosfolipidi e ioni calcio
Il fattore IX (fattore Christmas) è una glicoproteina di circa 57 kDa formata da una unica catena polipeptidica che può essere attivata dal fattore XIa (attraverso la via intrinseca, vedi Par.7.3.1g) o dal complesso costituito dal fattore tessutale, dal fattore VIIa e da ioni calcio (vedi Par. 7.3.1a). Nel corso dell’attivazione, il fattore IX viene trasformato prima in un prodotto intermedio, contenente una catena pesante e una catena leggera legate da un ponte disolfuro, poi nel fattore IXa, che si origina per distacco di un frammento peptidico dalla catena pesante.
Il fattore VIII (fattore antiemofilico) costituisce la porzione procoagulante del cosidetto "complesso del fattore VIII", che esso forma con una componente ad alto peso molecolare (proteina correlata al fattore VIII o fattore von Willebrand). Il fattore VIII, una glicoproteina costituita da una catena leggera di 80 kDa e da una catena pesante con una massa molecolare variabile da 90 a 200 kDa, è attivato in fattore VIIIa dalla trombina presente nel plasma. Il fattore von Willebrand è un multimero formato da identiche subunità glicoproteiche che svolge il ruolo di vettore del fattore VIII ed interviene nell’emostasi permettendo l’adesione delle piastrine alla parete vasale.
Il fattore IXa, il fattore VIIIa e gli ioni calcio formano un complesso in presenza dei fosfolipidi forniti direttamente dal fattore tessutale, che interviene come substrato essenziale per l’interazione dei singoli fattori della coagulazione. Questo complesso costituisce una via alternativa per l’attivazione del fattore X in fattore Xa.
7.3.1c. Formazione del fattore Xa
Entrambi i complessi esaminati nei Par. 7.3.1a e Par. 7.3.1b hanno la capacità di trasformare il fattore X in fattore Xa. Questa è una fase essenziale del processo di attivazione della coagulazione in quanto in essa confluiscono sia la sequenza principale, attivata dal fattore tessutale attraverso la via estrinseca, sia la sequenza che è in rapporto con la via intrinseca attraverso il fattore IXa.
Il fattore X (fattore Stuart) è una glicoproteina di 59 kDa formata da due catene poliptidiche unite da ponti disolfuro. La trasformazione del fattore X in Xa è caratterizzata dalla scissione di un legame arginina-isoleucina posto nella catena pesante con formazione di piccolo frammento e del fattore Xaα. Quest’ultimo viene ulteriormente scisso a livello di un legame arginina-glicina con formazione del fattore attivato definitivo (fattore Xaβ). I fattori Xaα e Xaβ mostrano simile attività biologica.
7.3.1d. Formazione del complesso costituito da fattore Xa, fattore Va, fosfolipidi e ioni calcio
Il fattore Xa entra a far parte di un complesso che utilizza come cofattore essenziale il fattore Va in presenza di fosfolipidi e ioni calcio. Il fattore V (proaccelerina) è una glicoproteina di 250-300 kDa costituita da almeno due catene polipeptidiche ed è attivato in fattore Va dalla trombina presente nel plasma. Il complesso costituito dal fattore Xa, dal fattore Va, dai fosfolipidi e da ioni calcio viene chiamato protrombinasi in quanto permette la trasformazione della protrombina in trombina.
7.3.1e. Formazione della trombina
La protrombina (fattore II) è una glicoproteina di 72 kDa formata da una unica catena polipeptidica. La trasformazione in trombina avviene attraverso una serie di reazioni catalizzate dal complesso costituito dal fattore Xa, dal fattore Va, dai fosfolipidi e dagli ioni calcio (Fig. 7.11). Il fattore Xa scinde la protrombina in due parti: una contenente i frammenti 1 e 2, l’altra costituita dalla pretrombina 2 (o prodotto intermedio II). La trombina deriva dalla pretrombina 2 quando questa viene scissa dal fattore Xa in due polipeptidi che rimangono tuttavia uniti da un ponte disulfuro. Il legame della trombina ai fosfolipidi di superficie è condizionato dalla presenza della proteina Z, una glicoproteina di 62 kDa.
La trombina è una proteasi a serina capace di agire sul fibrinogeno, sui fattori V, VII, VIII, XI e XIII, sulle piastrine, nonché sulla protrombina stessa. Infatti la trombina può idrolizzare la protrombina e staccare da essa il frammento 1. La parte di protrombina priva del frammento 1 è denominata pretrombina 1. Questa continua ad essere attaccabile dal fattore Xa, ma con maggiore difficoltà in quanto l’integrità del frammento 1-2 è necessaria per una rapida trasformazione della protrombina in trombina.
|
|
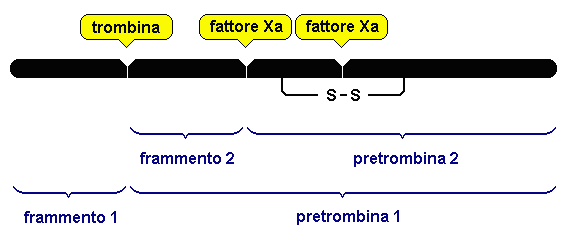
7.3.1f. Formazione della fibrina
L’ultima fase del processo coagulativo consiste nella trasformazione del
fibrinogeno in fibrina. Il fibrinogeno è una glicoproteina di 332 kDa costituita
da due monomeri, ciascuno dei quali è formato, a sua volta, da tre catene polipeptidiche denominate
α (63 kDa), β
(56 kDa) e γ (47 kDa)
![]() . Le catene sono tenute insieme
da ponti disolfuro. La molecola ha una forma allungata ed è caratterizzata
dalla presenza di tre noduli con una struttura globulare. Il nodulo posto al
centro della molecola (dominio E) contiene le porzioni aminoterminali delle sei catene,
mentre nei noduli posti alle due estremità (domini D) sono presenti
le regioni carbossiterminali delle catene β e γ. La porzione carbossiterminale delle catene
α si ripiega su se stessa ed interagisce con il
dominio E.
. Le catene sono tenute insieme
da ponti disolfuro. La molecola ha una forma allungata ed è caratterizzata
dalla presenza di tre noduli con una struttura globulare. Il nodulo posto al
centro della molecola (dominio E) contiene le porzioni aminoterminali delle sei catene,
mentre nei noduli posti alle due estremità (domini D) sono presenti
le regioni carbossiterminali delle catene β e γ. La porzione carbossiterminale delle catene
α si ripiega su se stessa ed interagisce con il
dominio E.
Le molecole di fibrinogeno sono trasformate in monomeri di fibrina dalla trombina che stacca dal versante aminoterminale delle due catene α e β due coppie di frammenti peptidici denominati rispettivamente fibrinopeptidi A e B. L’allontanamento dei fibrinopeptidi espone nuovi tratti aminoterminali sulle catene α e β nel dominio E. Questi sono capaci di interagire con i corrispondenti siti di legame presenti nei domini D. La polimerizzazione della struttura porta alla formazione di protofibrille tenute insieme da legami reversibili, facilmente dissociabili in soluzione di urea concentrata o in acido monocloroacetico.
La trasformazione della fibrina solubile nella forma insolubile avviene per intervento del fattore XIII (o fattore stabilizzante la fibrina), una proteina di circa 310 kDa formata da due catene A (75 kDa ciascuna) e due catene B (80 kDa ciascuna). Questo fattore, attivato dalla fibrina, catalizza le reazioni di transamidazione tra le catene γ e le catene α della fibrina, che danno luogo a dei legami covalenti tra i gruppi γ-carbossilici dei residui di acido glutammico di un monomero e i gruppi ε-aminici dei residui di lisina di un altro monomero. La fibrina così stabilizzata risulta più resistente in vivo all’azione fibrinolitica della plasmina.
7.3.1g. Attivazione da contatto
Con il termine "fattori di contatto" sono indicate quelle proteine che intervengono durante la prima fase di attivazione della via intrinseca. Queste proteine sono il fattore XII, il fattore XI, la precallicreina e il chininogeno ad alto peso molecolare. La presenza di tutti e quattro i fattori di contatto è necessaria per una ottimale attivazione del processo coagulativo in vitro. Per quanto riguarda le situazioni in vivo, si ritiene che l’attivazione da contatto possa svolgere un ruolo importante soprattutto in alcuni stati patologici, come nella coagulazione intravasale disseminata, nelle sepsi e negli stati di shock. Non è invece ancora del tutto noto il ruolo dei fattori di contatto in condizioni fisiologiche. E’ stato comunque visto che la carenza assoluta di fattore XII, di precallicreina o di chininogeno ad alto peso molecolare non dà luogo a manifestazioni emorragiche, mentre i soggetti omozigoti o eterozigoti per un deficit di fattore XI possono presentare una sindrome emorragica di varia gravità. Ciò suggerisce che, indipendentemente dall’attivazione del fattore XII, il fattore XI può svolgere un ruolo attivo nel processo coagulativo, in particolare quando è presente un inibitore specifico della via estrinseca (vedi Par. 7.3.1h), quando l’integrità vasale è severamente compromessa o quando i processi fibrinolitici sono particolarmente attivi.
Il fattore XII (fattore Hageman) è costituito da una singola catena polipeptidica di 80 kDa. Può essere attivato da enzimi proteolitici (callicreina, plasmina, fattore XIa, tripsina), dal contatto con superfici di vetro o da altre sostanze (acido ellagico, omocisteina, acidi grassi). La trasformazione nella forma attiva (fattore XIIa) avviene per idrolisi di un legame arginina-valina e formazione di una catena pesante, con siti di legame per superfici a carica negativa, e di una catena leggera che si comporta da proteasi a serina. Il fattore XIIa può attivare il fattore XI, il fattore VII, la precallicreina e l’attivatore tessutale del plasminogeno.
Il fattore XI (antecedente tromboplastinico plasmatico) è una proteina di 180 kDa formata da due catene identiche, che circola nel plasma legata al chininogeno ad alto peso molecolare. Può essere attivato dal fattore XIIa, dalla trombina e dalla tripsina mediante l’idrolisi di un legame arginina-isoleucina e formazione di un tetramero contenente due catene pesanti e due catene leggere. Il fattore XIa che così si forma può attivare il fattore IX in presenza di ioni calcio (vedi Par. 7.3.1b).
La precallicreina (fattore Fletcher) è formata da una singola catena polipeptidica di 83 kDa. Può essere attivata a callicreina
![]() dal fattore XIIa e
dalla stessa callicreina, già presente nel mezzo, mediante l’idrolisi di un legame arginina-isoleucina e formazione di un dimero
costituito da una catena pesante,
contenente il sito di legame per il chininogeno ad alto peso molecolare, e da una
catena leggera, contenente il sito catalitico. La callicreina agisce di concerto
con il fattore XIIa ed è capace di attivare il plasminogeno, il complemento, il
sistema delle chinine e il sistema renina-angiotensina, trasformando la
prorenina in renina.
dal fattore XIIa e
dalla stessa callicreina, già presente nel mezzo, mediante l’idrolisi di un legame arginina-isoleucina e formazione di un dimero
costituito da una catena pesante,
contenente il sito di legame per il chininogeno ad alto peso molecolare, e da una
catena leggera, contenente il sito catalitico. La callicreina agisce di concerto
con il fattore XIIa ed è capace di attivare il plasminogeno, il complemento, il
sistema delle chinine e il sistema renina-angiotensina, trasformando la
prorenina in renina.
Il chininogeno ad alto peso molecolare (HMW-K, high-molecular weight
kininogen) forma nel plasma dei complessi bimolecolari con il fattore XI e la precallicreina. Nella sua forma attiva priva
della componente costituita dalla bradichinina,
è formato da una singola catena glicoproteica di 120 kDa
![]() .
.
Quando il fattore XII viene a contatto con una superficie carica negativamente, subisce un cambiamento conformazionale che lo rende suscettibile all’azione proteolitica della callicreina e della plasmina. Si verifica in tal modo una reciproca attivazione tra fattore XII e precallicreina con formazione di fattore XIIa e callicreina. La callicrena stacca la bradichinina dal chininogeno ad alto peso molecolare, si lega a quest’ultimo ed abbandona con esso la superficie carica negativamente, permettendo così al fattore XI di interagire con il fattore XIIa e subirne l’attivazione. Il fattore XIa rimane adeso alla superficie carica negativamente e dà inizio alla cascata di reazioni che fanno parte della via intrinseca del processo coagulativo.
7.3.1h. Inibitori fisiologici della coagulazione
Le reazioni della cascata coagulativa sono controllate da meccanismi aspecifici e specifici di inibizione fisiologica. I meccanismi aspecifici sono costituiti innanzitutto dal normale flusso ematico che evita un possibile incremento locale dei fattori attivati e frammenta eventuali piccoli coaguli di fibrina. Gli inibitori specifici della coagulazione sono rappresentati dall’antitrombina III, dalla proteina C anticoagulante, dalla proteina S, dall’inibitore specifico della via estrinseca e dal cofattore eparinico II.
L’antitrombina III (o cofattore eparinico I) è una α2-globulina di 63 kDa che ha la proprietà di inibire diverse proteasi a serina costituenti alcuni fattori della cascata coagulativa, come la trombina, la callicreina ed i fattori IXa, Xa e XIa. Essa infatti contiene nel suo centro attivo un residuo di arginina che, legandosi al residuo di serina degli enzimi proteolitici, ne determina l’inattivazione. La reazione è favorita dall’eparina che interagisce con i residui di lisina dell’antitrombina III, rendendo immediatamente disponibile il centro attivo di questa antiproteasi (vedi Par.1.5.1d). L’importanza dell’antitrombina III è chiaramente documentata dalla grave sindrome trombotica presente nei soggetti con deficit di questa proteina e dall’efficacia della profilassi eparinica sull’insorgenza delle trombosi venose profonde.
La proteina C anticoagulante è presente nel plasma in una forma inattiva. Questa proteina è attivata dalla trombina legata ad un recettore endoteliale (trombomodulina) ed è così trasformata in una proteasi a serina (APC, activated protein C) capace di degradare ed inattivare i fattori VIIIa e Va della cascata coagulativa in presenza di proteina S, che agisce da cofattore non enzimatico. La proteina S è trasportata nel plasma per il 60% dalla proteina che lega la frazione C4b del complemento, ma solo la frazione libera della proteina S è quella funzionalmente attiva. Anche il fattore V agisce come cofattore dell’APC in quanto coopera con la proteina S nel potenziare la degradazione dei fattori VIIIa e Va.
L’inibitore specifico della via estrinseca (TFPI, tissue factor pathway inhibitor, o LACI, lipoprotein associated coagulation inhibitor, o EPI, extrinsic pathway inhibitor) è una proteina veicolata in gran parte dalle lipoproteine LDL, HDL e Lp(a) e per il 10% dalle piastrine, dalle quali è liberata a seguito di una stimolazione con la trombina. Questo inibitore forma un complesso quaternario con il fattore tessutale, il fattore VIIa e il fattore Xa, inattivando questi fattori.
Il cofattore eparinico II svolge un ruolo fisiologico ancora non bene definito inattivando la trombina, ma non il fattore Xa.
7.3.2. Fibrinolisi
Il sistema fibrinolitico ha lo scopo di limitare un eventuale danno ai tessuti che può derivare da una eccessiva deposizione di fibrina e dall’occlusione dei vasi. Come la cascata coagulativa, anche il sistema fibrinolitico è costituito da una serie di proteasi e di inibitori specifici. Questa cascata di reazioni enzimatiche porta alla trasformazione del plasminogeno in plasmina, una proteasi a serina capace di degradare la fibrina in frammenti solubili (FDP, fibrin degradation products). Oltre a solubilizzare la fibrina, la plasmina è capace anche di degradare il fibrinogeno ed altre proteine, come ad esempio i fattori V e VII della cascata coagulativa, alcune componenti del complemento, l’ACTH, l’ormone somatotropo e il glucagone.
Il plasminogeno è una proteina costituita da una singola catena di aminoacidi stabilizzata da 24 ponti disolfuro (Fig. 7.12). La forma nativa del plasminogeno (88
kDa) possiede in posizione N-terminale un residuo di acido glutammico ed è
pertanto definita glu-plasminogeno per distinguerla dalle altre isoforme
presenti nel plasma in minore quantità. Quest’ultime forme (definite nel loro
complesso lys-plasminogeno) derivano dal glu-plasminogeno per idrolisi del
legame tra i residui di lisina-77, lisina-78 o arginina-68 e residui
aminoacidici successivi. La molecola di plasminogeno è caratterizzata dalla
presenza di 5 strutture molto simili tra loro, chiamate kringle (dal nome
del dolce danese del quale ricordano la struttura) ed identificate con le
sigle da K1 a K5. Ciascun kringle è costituito da circa 80 residui
aminoacidici ed è stabilizzato da 3 ponti disolfuro
![]() . La regione con attività proteasica è presente nella porzione
carbossi-terminale della molecola ed è attivata a seguito dell’idrolisi del legame peptidico tra l’arginina-560 e la valina-561. L’attivazione, che porta alla
trasformazione del plasminogeno in plasmina, determina la scissione della catena
polipeptidica in due frammenti che rimangono tuttavia uniti da un ponte
disolfuro. Il lys-plasminogeno, presente soprattutto sulla superficie delle
cellule, è attivato a plasmina ad una velocità 10-20 volte superiore rispetto al
glu-plasminogeno e si lega più saldamente alla fibrina ed ai recettori
cellulari.
. La regione con attività proteasica è presente nella porzione
carbossi-terminale della molecola ed è attivata a seguito dell’idrolisi del legame peptidico tra l’arginina-560 e la valina-561. L’attivazione, che porta alla
trasformazione del plasminogeno in plasmina, determina la scissione della catena
polipeptidica in due frammenti che rimangono tuttavia uniti da un ponte
disolfuro. Il lys-plasminogeno, presente soprattutto sulla superficie delle
cellule, è attivato a plasmina ad una velocità 10-20 volte superiore rispetto al
glu-plasminogeno e si lega più saldamente alla fibrina ed ai recettori
cellulari.
|
|
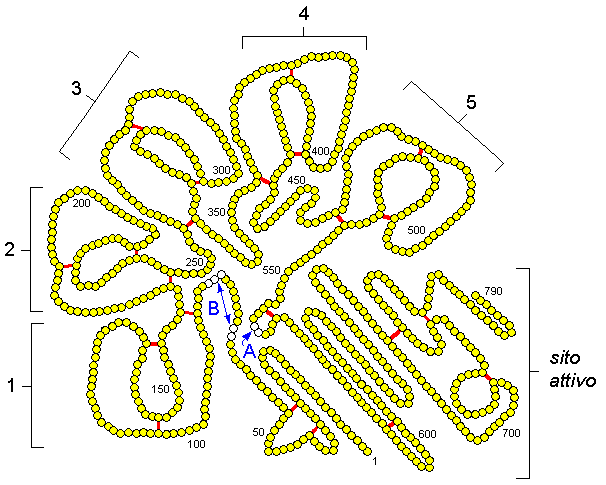
7.3.2a. Attivatori della fibrinolisi
L’attivazione del plasminogeno in plasmina può essere provocata da sostanze
esogene o da sostanze fisiologiche. Al primo gruppo appartengono diversi
prodotti somministrati come farmaci trombolitici o fibrinolitici nel trattamento
precoce delle trombosi
![]() . Al secondo gruppo appartengono l’attivatore
tessutale del plasminogeno (t-PA, tissue plasminogen activator), l’urochinasi
(u-PA, urokinase-type plasminogen activator) e, in minor misura, la
callicreina, il fattore XIa e il fattore XIIa.
. Al secondo gruppo appartengono l’attivatore
tessutale del plasminogeno (t-PA, tissue plasminogen activator), l’urochinasi
(u-PA, urokinase-type plasminogen activator) e, in minor misura, la
callicreina, il fattore XIa e il fattore XIIa.
L’attivatore tessutale del plasminogeno (t-PA) è sintetizzato dalle cellule endoteliali. La sua increzione in circolo può essere indotta diverse sostanze (trombina, istamina, epinefrina). La proteina (72kDa) ha una struttura omologa a quella del plasminogeno e ad altre proteine. Iniziando dalla porzione aminoterminale, si trova un dominio omologo ad un simile segmento presente nella fibronectina (finger domain), quindi un dominio simile all’epidermal growth factor, due strutture tipo kringle, ed infine il sito attivo tipico delle proteasi a serina. L’attivatore tessutale del plasminogeno è scisso nel plasma in due frammenti che rimangono uniti da un ponte disolfuro. La proteina nativa e quella scissa in due frammenti presentano proprietà catalitiche simili ed entrambe sono capaci di scindere il plasminogeno a plasmina. L’attività catalitica di entrambe le forme dell’attivatore è fortemente potenziata dalla fibrina che viene legata mediante il finger domain e il kringle-2 . Questo kringle è utilizzato anche per il legame dell’attivatore tessutale del plasminogeno all’inibitore PAI-1 (vedi Par. 7.3.2b).
L’urochinasi nativa è inattiva (prourochinasi, scu-PA, single chain u-PA) ed è costituita da una singola catena aminoacidica di 54 kDa contenente un dominio simile all’epidermal growth factor, un singolo kringle e il sito catalitico proprio delle proteasi a serina. La prourochinasi è trasformata nella forma attiva (urochinasi, u-PA) dalla plasmina o dalla callicreina che tagliano la struttura polipeptidica in due catene. L’urochinasi attivata può essere nuovamente attaccata dalla plasmina, perdere così un frammento di 135 residui e trasformarsi in una isoforma a più basso peso molecolare. Entrambe le isoforme ad alto e basso peso molecolare sono capaci di attivare il plasminogeno a plasmina, ma soltanto l’isoforma ad alto peso molecolare è capace di legarsi ad uno specifico recettore (u-PAR) espresso da numerosi tipi di cellule.
La callicreina ed i fattori XIa e XIIa contribuiscono solo in parte all’attivazione del plasminogeno e non sono capaci di supplire ad una carenza dell’attivatore tessutale del plasminogeno o dell’urochinasi.
7.3.2b. Inibitori della fibrinolisi
La plasmina è neutralizzata dalle antiproteasi presenti in circolo, in particolare dalla α2-antiplasmina (a2-AP) e dalla α2-macroglobulina (vedi Par. 8.3.3). L’α2-antiplasmina è una serpina di 70 kDa, prodotta principalmente dal fegato e presente in circolo alla concentrazione di circa 70 μg/mL con una vita media di circa 2,5 giorni. Il fattore XIII della coagulazione lega l’α2-antiplasmina alla catena α della fibrina, rendendo in tal modo la fibrina stessa più resistente alla degradazione operata dalla plasmina. L’α2-macroglobulina, che non appartiene al gruppo delle serpine, inibisce la plasmina meno efficacemente, formando con essa legami di tipo non-covalente.
L’azione degli attivatori del plasminogeno è neutralizzata da specifici inibitori (PAI, plasminogen activator inhibitors). Il PAI-1 (52 kDa) è sintetizzato nelle cellule muscolari lisce, negli adipociti e nei megacariociti ed è quindi presente nei granuli a delle piastrine. La sua sintesi nelle cellule endoteliali ed in numerosi altri tipi di cellule è fortemente stimolata da un’ampia gamma di mediatori dell’infiammazione e dalle citochine. Nel plasma è presente in forma attiva ad una concentrazione media di 50 ng/mL, ma decade rapidamente (la vita media è di circa 2 ore) per una modificazione conformazionale della sua struttura. La molecola è stabilizzata dalla vitronectina, una glicoproteina con proprietà adesive che serve a localizzare questo inibitore in specifiche regioni della matrice extracellulare. Il PAI-2 (47 kDa) è stato isolato originalmente nella placenta ed è presente nel plasma a livelli apprezzabili solo durante la gravidanza. Il PAI-3 (o inibitore della proteina C) viene sintetizzato dal tessuto renale e si trova nelle urine. Un ulteriore inibitore degli attivatori del plasminogeno è il PAI-4 (nexina I e II) che è capace di inibire anche la plasmina, la trombina, la tripsina e il fattore Xa.
Un altro inibitore della fibrinolisi è la carbossipeptidasi B (TAFI, thrombin activated fibrinolysis inhibitor) che è attivata nel corso della cascata coagulativa dalla trombina. La carbossipeptidasi B idrolizza i residui terminali di lisina presenti nella fibrina, impedendo in tal modo il legame del plasminogeno su di essa.
7.3.3. Alterazioni della funzione emostatica
Alterazioni congenite o acquisite dei meccanismi che regolano i processi di emocoagulazione e fibrinolisi possono portare a manifestazioni emorragiche e/o determinare la formazione di trombi.
7.3.3a. Diatesi emorragiche
L’emofilia è una malattia congenita legata al cromosoma X e caratterizzata da tendenza ad eccessivo sanguinamento. L’emofilia A è causata da mutazioni del gene che codifica per il fattore VIII, mentre l’emofilia B, più rara rispetto la precedente, è dovuta a mutazioni del gene che codifica per il fattore IX (vedi Par. 7.3.1b). Le mutazioni possono determinare la pressoché totale assenza di questi fattori nel sangue e, in tal caso, dare luogo a un quadro di emofilia grave (attività residua < 1%), oppure possono causare deficit parziali, con quadri di emofilia moderata (attività residua pari a 1-5% ) o lieve (attività residua pari a 6-35%) . Le portatrici della mutazione trasmettono la malattia, ma hanno generalmente valori plasmatici del fattore interessato inferiori solo di poco rispetto al normale (la differenza non è di norma evidenziabile all’esame del tempo di tromboplastina parziale, vedi Par. 7.3.4b) e non sviluppano perciò la malattia. Una alterazione della funzione emostatica può tuttavia manifestarsi anche nelle donne, se affette da una ineguale inattivazione o da una parziale emizigosi del cromosomna X. I pazienti sotto terapia sostitutiva possono frequentemente sviluppare alloanticorpi capaci di neutralizzare il fattore che viene loro somministrato.
Autoanticorpi contro il fattore VIII possono comparire anche in persone non
portatrici di una specifica mutazione sul gene che codifica questa proteina. Gli
autoanticorpi contro il fattore VIII possono determinare la comparsa di una
forma acquisita di emofilia; questa può essere idiopatica o associata a
collagenopatie o rappresentare una reazione abnorme a farmaci (ad esempio,
pennicillina). Un deficit parziale di fattore di fattore VIII è
riscontrabile nella malattia di von Willebrand e nel
deficit combinato di
fattore V e VIII, che è dovuto ad un raro difetto autosomico recessivo di una
proteina "chaperone"
![]() . Un deficit
combinato di fattore IX con i fattori VII e X, le proteine C e S e la
protrombina può essere causato da un difetto di γ-glutamilcarbossilasi
o di vitamina K (vedi Par. 7.3.1).
. Un deficit
combinato di fattore IX con i fattori VII e X, le proteine C e S e la
protrombina può essere causato da un difetto di γ-glutamilcarbossilasi
o di vitamina K (vedi Par. 7.3.1).
La malattia di von Willebrand è una malattia emorragica che viene trasmessa come carattere autosomico dominante a penetranza variabile ed è dovuta a carenza dell’omonimo fattore (vedi Par. 7.3.1b). Poiché la malattia si accompagna sempre a bassi livelli di fattore VIII, essa prende il nome di pseudoemofilia. In questo caso, tuttavia, il deficit di fattore VIII non è dovuto ad un difetto di sintesi, ma alla mancanza della funzione di trasporto e di stabilizzazione che il fattore di von Willebrand esercita normalmente sul fattore VIII.
Disordini riguardanti il fibrinogeno possono dare luogo a manifestazioni emorragiche. L’afibrinogenemia è un difetto ereditario autosomico recessivo con sintomatologia simile all’emofilia B, mentre l’ipofibrinogenemia e la disfibrinogenemia hanno manifestazioni cliniche meno gravi. Alcuni pazienti con disfibrinogenemia mostrano piuttosto una tendenza alla trombosi.
Altre rare forme di malattie emorragiche sono dovute a deficit congeniti di protrombina o dei fattori V, VII, X, XI o XIII. Deficit di fattore XII, precallicreina o chinogeno ad alto peso molecolare possono causare un allungamento del tempo di trombosplastina parziale (PTT, vedi Par. 7.3.4b) senza determinare tuttavia manifestazioni emorragiche.
La malattia emorragica può inoltre essere conseguenza di una trombocitopenia (vedi Par. 7.1.5a) o di una alterata funzione delle piastrine. La sindrome di Bernard-Soulier è un difetto autosomico recessivo che riguarda il recettore piastrinico (glicoproteina Ib) per il fattore di von Willebrand, mentre la trombastenia di Glanzmann è un difetto autosomico recessivo sul recettore glicoproteico IIb-IIIa che è coinvolto nel processo di aggregazione piastrinica. Numerosi farmaci possono infine alterare la funzionalità piastrinica (ad esempio, l’acido acetilsalicilico e l’alcool etilico).
7.3.3b. Trombofilia
Il termine "trombofilia" indica la tendenza a sviluppare trombi. Questi sono costituiti da una massa insolubile che si viene a formare nei vasi e nelle cavità cardiache a partire dalla fibrina (prodotta per azione della trombina sul fibrinogeno) e dalle piastrine aggregate. La trombosi venosa richiede la presenza di una stasi ematica, di un danno vascolare e di uno stato di ipercoagulabilità. Nella trombosi arteriosa l’intervento delle piastrine è molto più rilevante e può essere innescato da una interazione fra le piastrine stesse o, più frequentemente, dall’adesione di queste sulla parete vascolare, favorita dalla trombina. Gli stati di ipercoagulabilità possono essere primitivi (generalmente su base ereditaria) o secondari ad altre affezioni.
Le forme primitive possono derivare da un eccesso di fattori della coagulazione (elevati livelli di fattore II o VII, resistenza del fattore V alla inattivazione), da una diminuita disponibilità di inibitori fisiologici (deficit di antitrombina III, di proteina C o di proteina S) o da mutazioni riguardanti componenti del sistema fibrinolitico (elevati livelli di inibitore dell’attivatore del plasminogeno, vedi Par. 7.3.2b). La causa più comune di trombofilia su base ereditaria è una mutazione sul fattore V (variante Leida del fattore V) a livello del sito della catena aminoacidica dove avviene normalmente la sua inattivazione catalizzata dalla proteina C attivata (APC, vedi Par. 7.3.1h).
Altre situazioni in cui si può osservare una diminuzione dei livelli di
antitrombina III sono la gravidanza, alcune malattie epatiche, le sindromi
nefrotossiche, le trombosi diffuse, la coagulazione intravasale disseminata
(vedi Par. 7.3.3c) e il trattamento con l’enzima antitumorale L-asparaginasi
![]() . Un deficit di
proteina C può insorgere in pazienti sottoposti a terapie che prevedono l’uso di
anticoagulanti orali ad attività anti-vitamina K (vedi Par. 7.3.1). Associate a trombofilia sono
inoltre le sindromi da anticorpi antifosfolipidi. I due anticorpi appartenenti a
questo gruppo, che sono meglio
studiati, sono l’anticorpo anticardiolipina e l’anticoagulante tipo lupus
. Un deficit di
proteina C può insorgere in pazienti sottoposti a terapie che prevedono l’uso di
anticoagulanti orali ad attività anti-vitamina K (vedi Par. 7.3.1). Associate a trombofilia sono
inoltre le sindromi da anticorpi antifosfolipidi. I due anticorpi appartenenti a
questo gruppo, che sono meglio
studiati, sono l’anticorpo anticardiolipina e l’anticoagulante tipo lupus
![]() ; la presenza di
questi anticorpi è associata a trombosi venose o arteriose, a trombocitopenia e
ad aborto spontaneo ricorrente.
; la presenza di
questi anticorpi è associata a trombosi venose o arteriose, a trombocitopenia e
ad aborto spontaneo ricorrente.
Valori elevati di omocisteina nel sangue sono ritenuti fattori di rischio trombotico. I pazienti affetti da omocisteinuria
![]() , che hanno un
livello di omocisteina plasmatica 10 o 20 volte superiore alla norma, soffrono
di precoci danni vascolari, mentre i soggetti sani con valori di omocisteinemia
vicini ai valori superiori della norma hanno un rischio di infarto e di morte
cardiaca 3-4 volte più elevato rispetto agli altri soggetti sani
, che hanno un
livello di omocisteina plasmatica 10 o 20 volte superiore alla norma, soffrono
di precoci danni vascolari, mentre i soggetti sani con valori di omocisteinemia
vicini ai valori superiori della norma hanno un rischio di infarto e di morte
cardiaca 3-4 volte più elevato rispetto agli altri soggetti sani
![]() . Modesti aumenti
di omocisteinemia possono essere causati da vari fattori, come ad esempio
la particolare costituzione genetica, la carenza relativa di acido folico, di vitamina B6 o di vitamina B12,
l’insufficienza renale o la somministrazione di farmaci. Il
meccanismo che porta al processo aterotrombotico, non ancora completamente noto,
potrebbe coinvolgere la tiolazione e l’acetilazione di proteine plasmatiche ed
endoteliali nonché l’attivazione di processi infiammatori ed apoptotici.
. Modesti aumenti
di omocisteinemia possono essere causati da vari fattori, come ad esempio
la particolare costituzione genetica, la carenza relativa di acido folico, di vitamina B6 o di vitamina B12,
l’insufficienza renale o la somministrazione di farmaci. Il
meccanismo che porta al processo aterotrombotico, non ancora completamente noto,
potrebbe coinvolgere la tiolazione e l’acetilazione di proteine plasmatiche ed
endoteliali nonché l’attivazione di processi infiammatori ed apoptotici.
7.3.3c. Coagulazione intravasale disseminata
La coagulazione intravasale disseminata (coagulopatia da consumo, sindrome da defibrinazione) è una sindrome acquisita caratterizzata dalla contemporanea presenza di fenomeni emorragici e trombotici. Le situazioni scatenanti sono tutte quelle che causano l’ingresso in circolo del fattore tromboplastinico tessutale (vedi Par. 7.3.1a) e comprendono le ustioni gravi, i traumi o gli interventi chirurgici importanti (specialmente se a carico del cervello, polmoni o sistema genito-urinario), le tossiemie batteriche, le reazioni emolitiche trasfusionali e la leucemia acuta promielocitica. Le sepsi generalizzate e le gravi epatopatie possono causare una coagulazione intravasale disseminata sia in forma acuta che in una forma ad insorgenza graduale.
I fattori scatenanti determinano una massiccia attivazione della cascata coagulativa e, conseguentemente, la deposizione di fibrina all’interno dei piccoli vasi di molti tessuti ed organi. Ciò causa, a sua volta, una insufficiente ossigenazione tessutale, la deplezione dei fattori della coagulazione (in particolare dei fattori VIII e V, vedi Par. 7.3.1b e Par. 7.3.1d) e delle piastrine e l’attivazione secondaria del sistema fibrinolitico. Il fibrinogeno è diminuito ed incapace di coagulare, mentre in circolo sono presenti grandi quantità dei suoi prodotti di degradazione che inibiscono la formazione di fibrina. Queste alterazioni ematiche si sovrappongono a situazioni di stasi circolatoria, shock, ipovolemia ed aumentata permeabilità vasale, condizioni che compromettono ulteriormente la perfusione tessutale e rendono particolarmente difficile l’avvio di qualsiasi tentativo di compenso.
7.3.4. Metodi di determinazione
I test più importanti per interpretare le alterazioni della funzione emostatica sono il tempo di sanguinamento (secondo Ivy), il conteggio delle piastrine, il tempo di tromboplastina parziale attiva (aPTT, activated partial thromboplastin test), il tempo di protrombina (PT, prothrombin test), il dosaggio del fibrinogeno nel plasma.
Tra i test di seconda linea, più specifici e indagabili solo dopo una
attenta valutazione del tipo di alterazione della funzione emostatica, possiamo
ricordare lo studio dell’aggregabilità piastrinica e del tromboelastogramma e il tempo di trombina (TT, TCT, thrombin clotting time). Possono
infine essere dosati, generalmente mediante l’uso di anticorpi specifici, alcuni singoli
fattori, come ad esempio l’antitrombina III, la proteina C, la proteina S, il
plasminogeno, l’attivatore tessutale del plasminogeno (tPA), l’inibitore dell’attivatore
del plasminogeno (PAI-1), i prodotti di degradazione del fibrinogeno (FDP) e il
D-dimero
![]() .
.
Dopo che è stata formulata una ipotesi diagnostica sulla base delle alterazioni evidenziate dai test di screening, la conferma della diagnosi può essere raggiunta aggiungendo al plasma del paziente i diversi fattori, per verificare quale di essi sia in grado di correggere le anomalie riscontrate (prove di complementazione.)
7.3.4a. Metodi di esplorazione della fase piastrinica
Il tempo di sanguinamento (secondo Ivy) viene determinato misurando dopo quanto tempo cessa il sanguinamento da tre incisioni praticate sull’avambraccio ad una distanza di 1-2 cm fra loro, quando sul medesimo braccio è stata applicata con lo sfingomanometro una pressione di 40 mmHg.
Lo studio dell’aggregazione piastrinica viene eseguito con un metodo turbidimetrico basato sulle variazioni di trasmittanza che si ottengono in un plasma arricchito di piastrine dopo aggiunta di sostanze aggreganti (ADP, adrenalina, collageno, ristocetina). I parametri che sono presi in considerazione sono il tempo di latenza, la velocità di aggregazione e la percentuale di disaggregazione, dopo che è stato raggiunto il punto di massima aggregazione.
La tromboelastografia permette di indagare le caratteristiche fisiche del coagulo man mano che questo si forma. Lo strumento consiste in uno stantuffo collegato ad un filo di torsione ed immerso in una cuvetta termostatata a 37°C, contenente il campione di sangue. La cuvetta compie automaticamente dei movimenti periodici in senso orario ed antiorario con un breve arresto a fine corsa. Quando si forma il coagulo, i filamenti di fibrina fanno aderire la cuvetta al pistone che viene trascinato consensualmente facendo torcere il filo di torsione. Quest’ultimo movimento viene registrato in un tracciato, del quale vengono valutate la forma e l’ampiezza (Fig. 7.13).
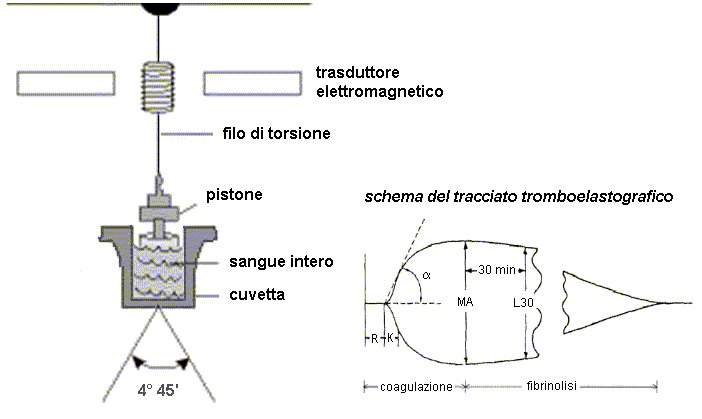
Fig. 7.13. Schema di un tromboelastografo e di un tracciato tromboelastografico. I parametri che sono valutati nel tromboelastogrammma sono il tempo di reazione (R), il tempo di formazione del trombo (K), la velocità di formazione del trombo (α), l’ampiezza massima del trombo (MA) e la percentuale di lisi a 30 minuti (L30).
7.3.4b. Tempo di tromboplastina parziale attivata (aPTT)
Il test viene eseguito su plasma, reso incoagulabile per aggiunta di
citrato, a
cui vengono addizionati calcio e degli agenti attivanti come il caolino e una
emulsione di fosfolipidi
![]() . Il calcio
aggiunto serve a rimpiazzare il calcio chelato dal citrato, il caolino ha la
funzione di innescare l’attivazione da contatto, mentre i fosfolipidi agiscono
da sostituti piastrinici fornendo una superficie adatta per le interazioni
enzima-substrato necessarie allo svolgimento del processo coagulativo.
. Il calcio
aggiunto serve a rimpiazzare il calcio chelato dal citrato, il caolino ha la
funzione di innescare l’attivazione da contatto, mentre i fosfolipidi agiscono
da sostituti piastrinici fornendo una superficie adatta per le interazioni
enzima-substrato necessarie allo svolgimento del processo coagulativo.
Il aPTT valuta l’efficacia della via intrinseca e della via comune della coagulazione ed è più sensibile rispetto al tempo di protrombina (PT, vedi Par. 7.3.4c) nell’individuare piccoli difetti di quest’ultima via. Il test misura la presenza dei fattori VIII, IX, XI e XII nonchè la presenza dei fattori X e V, della protrombina e del fibrinogeno. In generale, si osserva un allungamento del aPTT quando uno di questi fattori è presente a un livello inferiore al 30% del normale.
7.3.4c. Tempo di protrombina (PT)
Il test viene eseguito su plasma, reso incoagulabile per aggiunta di citrato, a cui vengono addizionati calcio e tromboplastina tessutale. Questi due reagenti sono in grado di innescare la cascata coagulativa attraverso l’attivazione del fattore X, senza richiedere l’intervento delle piastrine o dei fattori della via intrinseca.
Il test valuta l’efficacia della via estrinseca e della via comune della coagulazione e misura la presenza dei fattori VII, X e V, della protrombina e del fibrinogeno. Un allungamento isolato del PT, con tempo di tromboplastina parziale (PTT, vedi Par. 7.3.4b) normale, è caratteristico della carenza di fattore VII, mentre un allungamento sia del PT che del PTT si può verificare in molte situazioni, come nel caso di terapie con anticoagulanti orali, ipovitaminosi K, epatopatie o deficit della via comune della coagulazione.
Come tromboplastina tessutale è utilizzabile una preparazione estratta da cervello e opportunamente standardizzata, per permettere la formazione di un coagulo solido di fibrina nel plasma normale in un tempo compreso tra 11 e 13 secondi. Sono inoltre disponibili preparazioni di fattore tessutale ricombinante, che sono dotate di maggiore sensibilità. I valori di PT dipendono ovviamente dal tipo di tromboplastina tessutale, che è stata adoperata nel saggio, e sono pertanto soggetti ad ampie variazioni tra laboratorio e laboratorio. Per questo motivo, il risultato dell’analisi viene generalmente espresso in termini di rapporto protrombinico normalizzato (INR, international normalized ratio). Questo si basa sul raffronto fra i tempi di protrombina ottenuti sul sangue del paziente e su un controllo normale, tenendo conto altresì di un indice (ISI, international sensitivity index) assegnato dall’industria a ciascun batch di reagente. L’indice ISI, determinato per mezzo di una calibrazione con una preparazione di riferimento internazionale standard (il cui ISI è per definizione uguale ad 1), assume un valore tanto più grande rispetto all’unità quanto meno sensibile è il reattivo messo in commercio. Il valore dell’INR è calcolato attraverso la seguente formula:
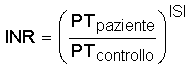
La determinazione del valore dell’ISI è molto critica in quanto questo termine è presente come esponente nella formula; di conseguenza piccoli errori nel calcolo dell’ISI possono alterare in modo rilevante il calcolo dell’INR. Pertanto il valore dell’INR è limitato alla standardizzazione delle terapie anticoagulanti, mentre esso non fornisce informazioni significative nei casi in cui il tempo di protrombina sia alterato a causa di fattori diversi (epatopatie, malassorbimento, ipovitaminosi K).
7.3.4d. Tempo di trombina (TT, TCT)
Il test viene eseguito su plasma, reso incoagulabile per aggiunta di citrato, a cui vengono addizionati calcio e trombina. Il test valuta quindi esclusivamente la parte terminale della via comune della coagulazione ed è utilizzabile per lo studio dell’interazione della trombina con il fibrinogeno.
Il tempo di trombina è aumentato in caso di deficit qualitativi o quantitativi del fibrinogeno (ossia quando i livelli plasmatici del fibrinogeno sono
inferiori a 100 mg/dL), nonché quando sono presenti anticoagulanti (ad esempio
l’eparina
![]() , vedi
Par. 1.5.1d) o
un eccesso di fibrinopeptidi (vedi Par. 7.3.1f). Tutte le
condizioni che causano un prolungamento del TT determinano ovviamente anche un
prolungamento dei tempi di tromboplastina parziale (PTT, vedi
Par. 7.3.4b) e di protrombina (PT, vedi Par. 7.3.4c).
, vedi
Par. 1.5.1d) o
un eccesso di fibrinopeptidi (vedi Par. 7.3.1f). Tutte le
condizioni che causano un prolungamento del TT determinano ovviamente anche un
prolungamento dei tempi di tromboplastina parziale (PTT, vedi
Par. 7.3.4b) e di protrombina (PT, vedi Par. 7.3.4c).
7.3.4e. Dosaggio del fibrinogeno
Il dosaggio del fibrinogeno viene eseguito su plasma che è stato reso incoagulabile per aggiunta di citrato ed è stato opportunamente diluito. La determinazione può essere effettuata misurando la quantità di proteina precipitabile per aggiunta di trombina oppure misurando la proteina che reagisce con anticorpi specifici, mediante tecniche di immunoprecipitazione. Nel caso di disfibrinogenemie (vedi Par. 7.3.3a) si possono osservare notevoli discrepanze tra la quantità di fibrinogeno in grado di coagulare e la quantità di proteina immunoreattiva.
7.3.5. Preparazione del campione ed intervalli di riferimento
Le analisi per la determinazione delle alterazioni della funzione emostatica sono generalmente eseguite su plasma reso incoagulabile per aggiunta di citrato di sodio, salvo particolari esigenze analitiche. Gli intervalli di riferimento dei principali test di coagulazione sono riportati in Tab. 7.IV.
|
Tab. 7.IV. Intervalli di riferimento dei principali test di coagulazione |
||
| Parametro | Valore di riferimento | |
| Tempo di sanguinamento | 1-6 min |
|
| Tempo di tromboplastina parziale attivata (aPTT) | 28-40 sec + 5 sec rispetto il controllo |
|
| Tempo di protrombina (PT) | 11-13 sec + 2 sec rispetto il controllo |
|
| Rapporto protrombinico normalizzato (INR) | 0,8-1,2 |
|
| Tempo di trombina (TT, TCT) | 10-15 sec <1,3 x valore di controllo |
|
| Fibrinogeno | 150-450 mg/dL | |
| aggiornamento: 22/02/12 |