|
|
Costantino Salerno |
|
|
Appunti di Biochimica Clinica |
|
|
5. SINTESI E CATABOLISMO DELL’EME
L’eme è il cofattore di numerose proteine che intervengono nel legame e nel trasporto dell’ossigeno (emoglobina e mioglobina), nel trasporto di elettroni (citocromi), nelle ossidazioni a funzione mista (citocromo P450), nella decomposizione del perossido d’idrogeno (perossidasi), nella sintesi del GMP ciclico (guanilato ciclasi) e nell’ossidazione del triptofano (triptofano pirrolasi), delle prostaglandine (prostaglandina endoperossido sintetasi) e dell’indolamina (indolamina diossigenasi).
5.1. COMPOSTI INTERMEDI NELLA BIOSINTESI DELL’EME
Come mostrato in Fig. 5.1, l’eme viene sintetizzato attraverso una sequenza di reazioni coinvolgenti otto enzimi: il primo (δ-aminolevulinico sintetasi) e gli ultimi tre enzimi (coproporfirinogeno ossidasi, protoporfirinogeno ossidasi e ferrochelatasi) sono situati nel mitocondrio, mentre gli enzimi intermedi della sequenza (δ-aminolevulinico deidratasi, porfobilinogeno deaminasi, uroporfirinogeno III cosintetasi e uroporfirinogeno decarbossilasi) si trovano nel citoplama. La biosintesi di una molecola di eme richiede 8 molecole di glicina e 8 molecole di succinil CoA. L’acido δ-aminolevulinico è il primo composto intermedio che si forma lungo questa via metabolica. Due molecole di acido δ-aminolevulinico si combinano tra loro in presenza della δ-aminolevulinico deidratasi a formare il porfobilinogeno, che è un monopirrolo, e 4 molecole di porfobilinogeno sono assemblate testa-coda ad opera della porfobilinogeno deaminasi a formare un tetrapirrolo lineare (idrossimetilbilano). La chiusura dell’anello e la formazione dell’uroporfirinogeno III sono catalizzate dalla cosintetasi che, operando un ribaltamento dell’ultimo dei nuclei pirrolici, impedisce l’accumulo degli isomeri tipo I, inutilizzabili per la sintesi dell’eme. L’uroporfirinogeno viene decarbossilato a coproporfirinogeno e quindi ossidato a protoporfirinogeno. I porfirinogeni si ossidano rapidamente all’aria formando le porfirine che sono i composti tetrapirrolici ciclici che si trovano normalmente nei campioni biologici. Una ossidasi specifica catalizza l’ossidazione del protoporfirinogeno IX a protoporfirina IX, che è substrato della ferrochelatasi, ultimo enzima della sequenza metabolica.
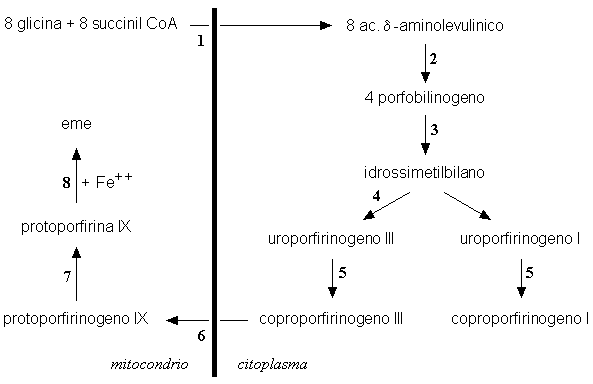
Figura 5.1. Biosintesi dell’eme. 1: δ-aminolevulinico sintetasi, 2: δ-aminolevulinico deidratasi, 3: porfobilinogeno deaminasi, 4: uroporfirinogeno III cosintetasi, 5: uroporfirinogeno decarbossilasi, 6: coproporfirinogeno ossidasi, 7: protoporfirinogeno ossidasi, 8: ferrochelatasi.
La sintesi dell’emoglobina nel midollo osseo richiede il consumo di circa 0,3 g di acido δ-aminolevulinico al giorno, mentre la sintesi dell’eme epatico (in gran parte impiegato nella formazione del citocromo P450 microsomiale) utilizza circa 0,05 g di acido δ-aminolevulinico al giorno. Quantità minori di acido δ-aminolevulinico intervengono nella sintesi dell’eme degli altri tessuti extraepatici.
La formazione dell’emoglobina è regolata mediante un
processo che passa attraverso la differenziazione delle cellule
eritroidi e che è associato all’aumento di tutti gli enzimi
della biosintesi dell’eme (in queste cellule la velocità di
sintesi dell’eme sembra dipendere in particolare dalla
disponibilità di ferrochelatasi). Nel fegato la biosintesi dell’eme
è invece regolata dall’attività della δ-aminolevulinico
sintetasi che è, a sua volta, influenzata da derivati ormonali (soprattutto
da metaboliti di ormoni steroidei aventi l’anello A saturo),
da farmaci o da fattori dietetici ![]() . Questi agiscono principalmente alterando la
concentrazione intracellulare dell’eme libero che,
attraverso un meccanismo di retroreazione negativa, inibisce sia
la sintesi della δ-aminolevulinico
sintetasi a livello trascrizionale e translazionale sia l’incorporazione
dell’enzima nel mitocondrio. La concentrazione
intracellulare dell’eme libero può ridursi a seguito di una
sua diminuita sintesi per inibizione della ferrochelatasi (ad
esempio, in conseguenza della somministrazione di antibiotici
come la griseofulvina) o a seguito di un suo aumentato consumo ad
opera dell’eme ossigenasi (la cui sintesi può essere
indotta, ad esempio, in condizioni di carenza alimentare) o per
la sintesi del citocromo P450 (ad esempio, in
conseguenza della somministrazione di anestetici come il
fenobarbital).
. Questi agiscono principalmente alterando la
concentrazione intracellulare dell’eme libero che,
attraverso un meccanismo di retroreazione negativa, inibisce sia
la sintesi della δ-aminolevulinico
sintetasi a livello trascrizionale e translazionale sia l’incorporazione
dell’enzima nel mitocondrio. La concentrazione
intracellulare dell’eme libero può ridursi a seguito di una
sua diminuita sintesi per inibizione della ferrochelatasi (ad
esempio, in conseguenza della somministrazione di antibiotici
come la griseofulvina) o a seguito di un suo aumentato consumo ad
opera dell’eme ossigenasi (la cui sintesi può essere
indotta, ad esempio, in condizioni di carenza alimentare) o per
la sintesi del citocromo P450 (ad esempio, in
conseguenza della somministrazione di anestetici come il
fenobarbital).
La solubilità delle porfirine in solventi acquosi diminuisce con il diminuire dei gruppi carbossilici dei sostituenti sull’anello tetrapirrolico. Così, l’uroporfirina (con 8 gruppi carbossilici) è la più solubile in acqua e viene escreta in gran parte con le urine, mentre la protoporfirina (con 2 gruppi carbossilici) è la meno solubile e viene eliminata solo con la bile. La coproporfirina con 4 gruppi carbossilici è escreta attraverso entrambe le vie. Oltre ai precedenti cataboliti, nelle urine possono essere presenti anche porfirine con cinque, sei e sette residui carbossilici derivanti da una decarbossilazione incompleta dei metaboliti intermedi della biosintesi dell’eme (Fig. 5.2).
Posizione |
||||||||
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
| Uroporfirina I | A |
P |
A |
P |
A |
P |
A |
P |
| Uroporfirina III | A |
P |
A |
P |
A |
P |
P |
A |
| Coproporfirina I | M |
P |
M |
P |
M |
P |
M |
P |
| Coproporfirina III | M |
P |
M |
P |
M |
P |
P |
M |
| Protoporfirina IX | M |
V |
M |
V |
M |
P |
P |
M |
Fig. 5.2. Porfirinogeni e porfirine
Ad eccezione della δ-aminolevulinico sintetasi, tutti gli enzimi che portano alla formazione dell’eme possono essere carenti e causare un aumento della concentrazione e dell’escrezione dei metaboliti intermedi della via biosintetica. Questi disordini biochimici sono distinti da alcuni Autori in porfirie (generalmente su base congenite), porfirinemie (per aumento della protoporfirina eritrocitaria in conseguenza di una carenza di ferro o di un avvelenamento da piombo) e porfirinurie (per un moderato aumento della coproporfirina urinaria). La maggior parte delle porfirie si trasmette come carattere autosomico dominante. Le eccezioni sono rappresentate dalla porfiria eritropoietica, dalla porfiria epatoeritropoietica e dalla carenza di δ-aminolevulinico deidratasi, che vengono trasmesse come malattie autosomiche recessive. La porfiria cutanea tarda può manifestarsi anche in forma sporadica ed essere scatenata dall’ingestione di alcool etilico, estrogeni o idrocarburi aromatici polialogenati o insorgere in forma epidemica per avvelenamento accidentale della popolazione (nel 1956 si verificarono 4000 casi in Turchia per immissione sul mercato di frumento trattato con esaclorobenzene). Il piombo ha effetti su diversi enzimi coinvolti nella sintesi dell’eme, inibendo la ferrochelatasi, la δ-aminolevulinico deidratasi, la porfobilinogeno deaminasi, la coproporfirinogeno ossidasi e, nei gravi stati di intossicazione, la uroporfirinogeno decarbossilasi.
In relazione ai principali siti di espressione del difetto
enzimatico, le porfirie sono classificate in eritropoietiche ed
epatiche (Tab. 5.I). Le manifestazioni
cliniche riguardano lesioni cutanee croniche da
fotosensibilizzazione per attivazione della perossidazione
lipidica e del complemento e manifestazioni acute neuroviscerali
forse per una diretta azione neurotossica dell’acido δ-aminolevulinico o per una insufficiente
attività della triptofano pirrolasi ![]() .
.
Tab. 5.I. Classificazione delle principali porfirie
Caratteristiche biochimiche |
||||||
| Classificazione | Deficit |
Sintomi |
Eritrociti |
Urine |
Feci |
|
| Porfirie
eritropoietiche
|
||||||
| Porfiria eritropoietica congenita | uroporfirinogeno cosintetasi |
fotosensibilizzazione |
uroporfirina |
uroporfirina |
coproporfirina |
|
| Protoporfiria eritropoietica | ferrochelatasi |
fotosensibilizzazione |
protoporfirina |
|
protoporfirina |
|
| Porfirie epatiche | ||||||
| Porfiria
acuta intermittente
|
porfobilinogeno deaminasi |
neuroviscerali |
δ.aminolevulinico |
|||
| Coproporfiria ereditaria | coproporfirinogeno ossidasi |
neuroviscerali |
δ.aminolevulinico |
|||
| Deficit di ALA deidratasi | δ.aminolevulinico deidratasi |
neuroviscerali |
protoporfirina |
δ.aminolevulinico |
|
|
| Porfiria
variegata
|
protoporfirinogeno ossidasi |
neuroviscerali |
δ.aminolevulinico |
coproporfirina |
||
| Porfiria
cutanea tarda
|
uroporfirinogeno decarbossilasi |
fotosensibilizzazione |
uroporfirina |
isocoproporfirina |
||
| Porfiria epatoeritropoietica | uroporfirinogeno decarbossilasi |
neuroviscerali |
protoporfirina |
uroporfirina |
isocoproporfirina |
|
5.1.1. Metodi di determinazione
La determinazione dell’acido δ-aminolevulinico e del porfobilinogeno richiede una preventiva modificazione chimica dell’analita generalmente attraverso una reazione colorimetrica. Il dosaggio delle porfirine non necessita invece di alcun processo di derivatizzazione in quanto queste sostanze assorbono luce nel visibile. Il loro spettro di assorbimento è caratterizzato da una banda principale attorno ai 400 nm (banda di Soret) e da quattro bande di intensità decrescente verso il rosso nella regione compresa fra 500 e 630 nm. Queste ultime bande di assorbimento si riducono a due in ambiente acido o in presenza di metalli. Inoltre, le porfirine, quando non sono chelate a metalli paramagnetici, sono intensamente fluorescenti.
5.1.1a. Determinazione dell’ac. δ-aminolevulinico e del porfobilinogeno
L’acido δ-aminolevulinico viene fatto reagire con un β-dichetone a formare un prodotto di condensazione contenente un nucleo pirrolico (ALA pirrolo). Questo, così come il porfobilinogeno, viene dosato dopo opportuna derivatizzazione. L’ALA pirrolo e il porfobilinogeno sono separabili fra loro e da eventuali altre sostanze interferenti mediante tecniche di purificazione che possono precedere o seguire le modificazioni chimiche a cui sono sottoposti.
(a) Formazione dell’ALA pirrolo
La reazione di condensazione fra il campione deproteinizzato e il β-dichetone (acetilacetone o etilacetoacetato) avviene a caldo (Fig. 5.3). La principale interferenza è dovuta ad un secondo aminochetone (l’aminoacetone) normalmente presente nei campioni biologici e derivante dalla ossidazione della treonina o dalla condensazione della glicina con l’acetil CoA.
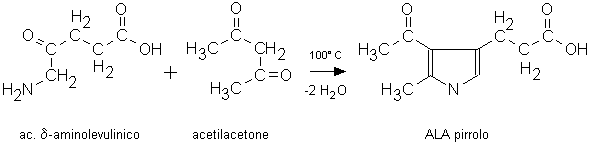
Fig. 5.3. Formazione dell’ALA pirrolo
(b) Derivatizzazione dei composti contenenti il nucleo pirrolico
L’ALA pirrolo e il porfobilinogeno sono fatti reagire con il reattivo di Ehrlich (p-dimetilaminobenzaldeide) in ambiente acido (Fig. 5.4). La reazione procede attraverso due tappe successive: prima si forma un prodotto di condensazione di colore rosso (dosabile spettrofotometricamente a 555 nm); in un secondo momento il composto rosso reagisce con un’altra molecola dell’analita formando un derivato incolore. Il prodotto colorato si mantiene per più tempo quando il reattivo di Ehrlich è preparato in acido perclorico (invece che in acido cloridrico). In queste condizioni, tuttavia, il reattivo di Ehrlich è più instabile. L’urobilinogeno, l’indolo, l’indacano, l’urea e l’acido ascorbico possono interferire nella reazione colorimetrica.
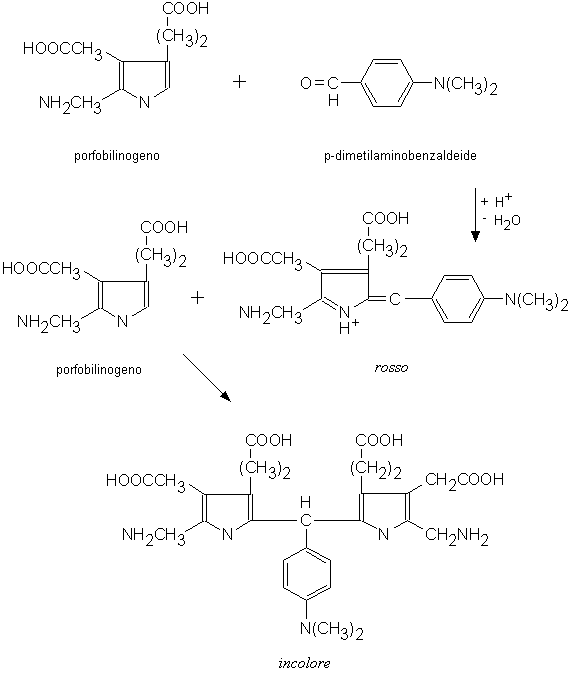
Fig. 5.4. Derivatizzazione del porfobilinogeno
Altri procedimenti prevedono la sililazione o la metilazione dell’ALA pirrolo e costituiscono tappe intermedie per la determinazione dell’acido δ-aminolevulinico mediante gas-cromatografia con rivelatore a ionizzazione di fiamma. L’analisi mediante gas-cromatografia ha una elevata sensibilità (1 ng/mL contro gli 0.5 μg/mL della metodica colorimetrica) ed è utilizzabile per la determinazione dell’acido δ-aminolevulinico nel siero.
(c) Tecniche di purificazione dell’addotto
L’addotto di colore rosso fra il porfobilinogeno e il reattivo di Ehrlich può essere purificato mediante estrazione in cloroformio dopo alcalinizzazione per aggiunta di acetato di sodio. Questo metodo (test di Watson-Schwartz), reso più specifico da una successiva estrazione delle sostanze interferenti residue in butano, è utilizzabile per evidenziare in modo non quantitativo un incremento di porfobilinogeno nelle urine. I metodi quantitativi, invece, prevedono di solito una separazione cromatografica degli analiti su resine a scambio ionico prima di iniziare i processi di derivatizzazione. Una volta formato l’ALA pirrolo, questo può essere ulteriormente purificato ripetendo la cromatografia a scambio ionico. In alternativa ai metodi cromatografici a bassa pressione, è possibile separare l’acido δ-aminolevulinico e il porfobilinogeno su HPLC. La determinazione quantitativa degli analiti è effettuata in tal caso dopo reazione colorimetrica in un reattore termostatato posto tra la colonna cromatografica e il rivelatore spettrofotometrico.
5.1.1b. Determinazione delle porfirine.
I porfirinogeni eventualmente presenti sono ossidati a porfirine con perossido di idrogeno. Le porfirine possono essere estratte da campioni debolmente acidificati mediante un solvente organico relativamente polare (di norma si usa una miscela acido acetico-etilacetato). Le porfirine sono quindi riportate in ambiente acquoso con acido cloridrico (3 M) e osservate alla luce ultravioletta, dove emettono una intensa fluorescenza rossastra.
Determinazioni quantitative possono essere ottenute mediante diverse tecniche. Un primo metodo consiste nella estrazione frazionata delle porfirine in solventi organici a un pH prossimo al loro punto isoelettrico (è possibile così separare l’uroporfirina dalla coproporfirina) e nella misura spettrofotometrica dell’estratto risolubilizzato in acido. In alternativa, è possibile adoperare un metodo cromatografico su strato sottile per separare le porfirine come tali o dopo una loro conversione in metil esteri mediante aggiunta di metanolo e acido solforico. Tecniche di cromatografia liquida ad alta pressione in fase inversa permettono una buona separazione di tutte le frazioni.
5.1.2. Preparazione del campione ed intervalli di riferimento
Il saggio può richiedere campioni di sangue, urine o feci (circa 1 g). La determinazione dell’acido δ-aminolevulinico e delle porfirine è eseguita preferibilmente sulle urine delle 24 ore. Nel caso del porfobilinogeno, è invece richiesto un campione di urine ottenuto durante o immediatamente dopo l’attacco acuto di dolore addominale; le urine devono essere raffreddate a temperatura ambiente prima di eseguire il test.
Una cattiva conservazione dei campioni può facilmente inficiare i risultati. Per la determinazione dell’acido δ-aminolevulinico è consigliabile raccogliere le urine in un recipiente contenente acido tartarico (0,5 mol/L) o acido benzoico (0,5 g/L). Nel caso delle porfirine la raccolta deve avvenire in presenza di bicarbonato di sodio (0,5 g/L). Le urine devono essere conservate nel congelatore e al buio.
L’intervallo di riferimento dell’acido δ-aminolevulinico è di 4–17 μg/L (30,5–130 nmol/L) nel plasma e di 25–100 μg/h (191–763 nmol/h) nelle urine. Nel caso di una intossicazione da piombo, questi valori salgono a 14-190 μg/L nel plasma ed a 83-2500 μg/h nelle urine. Per quanto riguarda il porfobilinogeno, il test può considerarsi negativo per concentrazioni urinarie dell’analita inferiori a 2 mg/L. La concentrazione delle porfirine deve essere inferiore a 600 μg/L nel sangue intero, a 200 μ/L nelle urine ed a 60 μg/g per peso secco di feci.
5.2. CATABOLITI DELL’EME
L’anello tetrapirrolico dell’eme è aperto a livello del ponte α-metenico (Fig. 5.5). La prima reazione è catalizzata dall’eme ossigenasi microsomiale, molto attiva nella milza, che porta alla formazione del α-ossieme. Questo composto viene a sua volta ossidato a biliverdina (biliverdina IXα) dall’ossigeno molecolare probabilmente mediante una reazione non-enzimatica che porta alla liberazione di monossido di carbonio. La biliverdina è convertita in bilirubina (bilirubina IXα) dalla biliverdina redutttasi, un enzima citosolico utilizzante NADH o NADPH come agenti riducenti. La struttura della bilirubina è stabilizzata da legami idrogeno fra i gruppi carbossilici dei residui propionici e gli anelli pirrolici esterni. La molecola, meno polare della biliverdina, è instabile in soluzioni acquose e tende a formare aggregati colloidali.
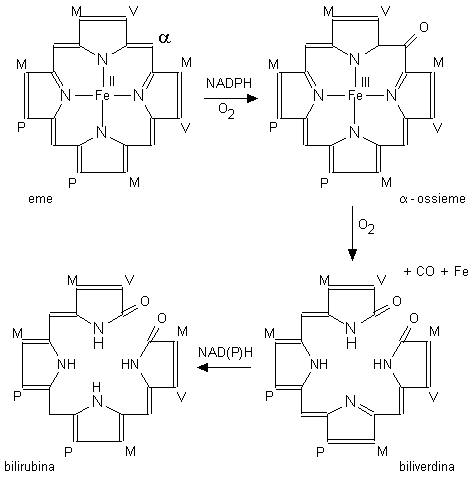
Fig. 5.5. Ossidazione dell’eme a bilirubina
Circa l’80% della bilirubina deriva dalla degradazione dell’eme liberato dalla lisi degli eritrociti senescenti. Una quota minore proviene dalle altre emoproteine. Una frazione ancora più limitata deriva dall’eritropoiesi "inefficace", che può tuttavia diventare una fonte importante di bilirubina in alcune malattie ematologiche come la talassemia e l’anemia perniciosa (vedi Par. 7.1.3a).
In circolo, la bilirubina forma due diversi complessi con l’albumina: (1) in condizioni normali, la bilirubina si lega quasi esclusivamente come dianione ad un sito ad alta affinità dell’albumina; (2) in condizioni di iperbilirubinemia accompagnata ad acidosi, la bilirubina non-ionizzata è unita ad un sito a più bassa affinità mediante un legame fortemente dipendente dal pH. Numerose sostanze possono interferire con il legame dell’albumina con la bilirubina. I sulfamidi, i farmaci antiinfiammatori e il mezzo di contrasto usato nelle colangiografie spostano competitivamente la bilirubina dall’albumina. Gli acidi grassi si comportano in maniera diversa a seconda della loro lunghezza: quelli a catena breve inibiscono il legame della bilirubina all’albumina, quelli a catena media lo facilitano, quelli a catena lunga sono privi di effetto.
La bilirubina è rapidamente rimossa dal circolo e incorporata negli epatociti mediante un meccanismo di trasporto saturabile ed inibibile competitivamente da altri anioni organici, come l’indocianina e la bromosulftaleina. All’interno dell’epatocita, la bilirubina si lega alla proteina Y (o ligandina, identificata con la GSH-transferasi B) e, in minor misura, alla proteina Z.
Una efficiente escrezione della bilirubina attraverso i canalicoli biliari richiede una sua conversione in un composto più polare. Ciò avviene principalmente mediante l’esterificazione di uno o di entrambi i gruppi propionilici con l’acido glucuronico attraverso la reazione catalizzata dalla UDP glucuronosiltransferasi (Fig. 5.6). L’enzima, che si trova nei microsomi, è attivato dalla UDP-N-acetilglucosamina e può accettare come donatori del gruppo glucoronile sia l’UDP-glucuronato che l’UDP-glucosio e l’UDP-xilosio. Poiché la bilirubina IXa è asimmetrica, i suoi derivati monoesterificati esistono come due isomeri distinti, a seconda quale dei due gruppi propionilici sia interessato al legame. Il trasporto della bilirubina coniugata dall’epatocita nella bile avviene attraverso una pompa ATP-dipendente specifica per la bilirubina e per altri anioni organici ed è facilitato dal gradiente elettrochimico di membrana.
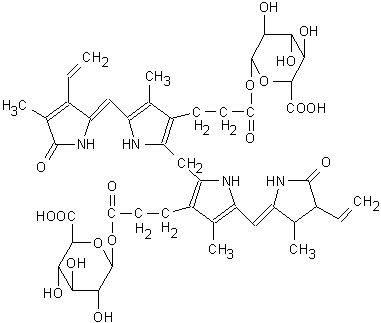
Fig. 5.6. Bilirubina diglucuronata
Nell’intestino, una parte della bilirubina è deconiugata dalle glucuronidasi batteriche e, ritornata così liposolubile, è riassorbita, ma la maggior parte è ossidata a urobilinogeno, che è ulteriormente metabolizzato ad altri pigmenti, soprattutto stercobilina, ed escreto. Una piccola parte dell’urobilinogeno viene riassorbita e va incontro ad una circolazione enteroepatica. Anche la bilirubina coniugata è legata all’albumina nel sangue circolante, ma con minore affinità (salvo una quota che forma invece un legame irreversibile), cosicché la parte non legata può essere filtrata dal glomerulo renale e la frazione che sfugge al riassorbimento tubulare può essere eliminata nell’urina.
Benché la produzione giornaliera di bilirubina sia inferiore a 500 mg, il fegato normale può arrivare a coniugarne più di 1500 mg al giorno. Quest’ampia riserva funzionale è una delle ragioni per cui la determinazione della bilirubinemia è un test di limitata sensibilità nelle epatopatie. Di grande rilevo clinico è invece la determinazione della frazione non coniugata della bilirubina nei casi di encefalopatia da ittero nucleare (kernittero). Questa alterazione si manifesta in neonati con alti livelli di bilirubina non coniugata o in giovani adulti con grave deficit ereditario di UDP-glucuronosiltransferasi. La bilirubina infatti inibisce la sintesi proteica, il metabolismo dei carboidrati e la respirazione cellulare nel cervello. Il kernittero si manifesta di solito fra il terzo e il sesto giorno di vita con ipotonia, iporiflessia, movimenti atetoidi ed opistotono. L’alterazione metabolica può evolvere portando a morte il paziente o dar luogo a perdita d’udito, paralisi dei nervi cranici, atetosi e ritardo mentale. Il danno neurologico è provocato dalla quota di bilirubina non coniugata che non è legata all’albumina (bilirubina libera), così che il rischio di kernittero aumenta con il diminuire della capacità legante residua dell’albumina. Si ritiene che l’immaturità della barriera emato-encefalica nel neonato possa contribuire al kernittero, ma non vi sono a tutt’oggi delle prove dirette in tal senso. La classificazione degli stati iperbilirubinemici è riportata in Tab. 5.II.
Tab. 5.II. Stati iperbilirubinemici.
| Iperproduzione di bilirubina | |||
| Itteri emolitici | |||
| Itteri da iperproduzione non emocateretica | |||
| Difetti funzionali dell’epatocita | |||
| congeniti | |||
| deficit di captazione (s. di Gilbert) | |||
| deficit di glucuronazione | |||
| transitoria (ittero neonatale) | |||
| permanente (s. di Crigler-Najjar tipo
I e II
|
|||
| deficit di trasporto ed
escrezione (s. di Dubin-Johnson, s. di Rotor
|
|||
| acquisiti | |||
| epatopatie acute e croniche | |||
| inibitori della
glucuronidasi (s. di Lucey
|
|||
| farmaci (novocaina) | |||
| Colestasi intraepatica | |||
| epatite colangiolitica | |||
| cirrosi biliare primitiva | |||
| atresia dei dotti | |||
| colangite sclerosante | |||
| Colestasi extraepatica | |||
| ab intrinseco (calcoli, neoplasie, parassitosi) | |||
| ab extrinseco (neoplasie, cisti, flogosi, etc.) | |||
La tradizionale classificazione dell’ittero in
preepatico, epatico e postepatico può essere ancora oggi utile.
Nell’ittero preepatico il carico di bilirubina che arriva al
fegato è aumentato generalmente per un processo emolitico in
atto dovuto a fragilità eritrocitaria (per esempio una
sferocitosi congenita) o ad una reazione immunitaria (ad esempio
una incompatibilità materno-fetale; vedi
Par. 7.1.3a). La bilirubina si trova
nella forma non coniugata e quindi non compare nell’urina
![]() . L’esame degli
indici ematologici è in genere sufficiente a consentire la
diagnosi, specie se fanno contrasto i test di funzionalità
epatica nei limiti della norma. L’analisi delle frazioni
coniugata e non coniugata assume in questi casi interesse quando
si voglia accertare se, in un paziente in cui sia già nota una
epatopatia cronica, l’emolisi possa essere una componente di
un eventuale peggioramento dell’ittero.
. L’esame degli
indici ematologici è in genere sufficiente a consentire la
diagnosi, specie se fanno contrasto i test di funzionalità
epatica nei limiti della norma. L’analisi delle frazioni
coniugata e non coniugata assume in questi casi interesse quando
si voglia accertare se, in un paziente in cui sia già nota una
epatopatia cronica, l’emolisi possa essere una componente di
un eventuale peggioramento dell’ittero.
La presenza di bilirubina nell’urina è sempre una manifestazione patologica e sta ad indicare una malattia del fegato (ittero epatocellulare) o delle vie biliari (ittero postepatico). La bilirubinuria può precedere una iperbilirubinemia e può quindi essere un indice assai sensibile di un danno epatico imminente. L’aumento della bilirubina nel plasma è soprattutto a carico della frazione coniugata. Se la quantità di pigmenti biliari che arriva all’intestino è ridotta (come nell’ittero ostruttivo), proporzionalmente diminuisce la quantità di urobilinogeno nell’urina. Se questo risulta assente ad una analisi ripetuta per più giorni, la causa più frequente è un carcinoma della testa del pancreas. La determinazione dell’urobilinogeno è tuttavia diventata obsoleta, sia per la complessità dell’analisi, sia per la disponibilità di indagini più affidabili ed accurate per la diagnosi di ostruzione delle vie biliari.
La bilirubina legata irreversibilmente all’albumina mediante un legame covalente (bilialbumina) costituisce una terza forma di bilirubina che può essere presente nel plasma. La bilirubina glucuronata è il precursore che dà origine a questo complesso. La bilialbumina compare nel plasma in tutte le condizioni che causano un accumulo del pigmento glucuronato, come l’ostruzione biliare, l’epatite, la cirrosi, le neoplasie epatiche e la sindrome di Dubin-Johnson e di Rotor. La formazione del complesso, che può costituire più del 90% della bilirubina totale, è dipendente dalla durata della colestasi, motivo per cui la percentuale di bilirubina legata irreversibilmente tende ad aumentare nel corso della malattia. Questa frazione di bilirubina non attraversa il filtro renale e possiede una emivita plasmatica simile all’albumina, spiegando la frequente assenza di bilirubinuria pur in presenza di elevati livelli di bilirubina coniugata nel plasma durante la fase di risoluzione di una patologia epatica ostruttiva. La bilialbumina non è invece dosabile nei soggetti normali e in quelli con iperbilirubinemia non coniugata, compresa la sindrome di Gilbert e di Crigler-Najjar.
5.2.1. Metodi di determinazione
I metodi per il dosaggio della bilirubina prevedono una sua determinazione spettrofotometrica diretta, l’uso di reazioni colorimetriche o la determinazione dell’analita dopo la sua purificazione. Ulteriori metodiche sono utilizzate per evidenziare la quota non legata all’albumina della bilirubina non glucuronata (bilirubina libera) e la capacità legante residua dell’albumina.
5.2.1a. Determinazione spettrofotometrica diretta
E’ possibile misurare direttamente allo spettrofotometro la concentrazione della bilirubina totale. Poiché alla lunghezza d’onda di massimo assorbimento della bilirubina (454 nm) anche l’ossiemoglobina assorbe la luce, è necessario correggere la misura per il contributo di quest’ultima sostanza. Per fare ciò, si sottrae alla misura a 454 nm l’assorbimento a 540 nm, poiché l’ossiemoglobina assorbe la luce in eguale misura a queste due lunghezze d’onda. Il metodo, tuttavia, non corregge per il contributo dato dai carotenoidi, che anche assorbono a 454 nm e possono dar luogo a misure falsamente elevate. Per questo motivo la determinazione spettrofotometrica diretta della bilirubina è raramente usata nell’adulto. Essa può essere invece adoperata nei neonati sotto i 3 mesi, che non assumono quantità di carotene sufficienti ad alterare il risultato del test. Una determinazione spettrofotometrica diretta del pigmento può essere fatta anche per stimare il livello di bilirubina nel liquido amniotico, come indice di un processo morboso nel feto.
5.2.1b. Reazioni colorimetriche
Molti dei metodi in uso per la determinazione della bilirubina nei liquidi biologici sono ancora oggi basati sulla diazo-reazione descritta da van den Bergh e Snapper nel 1913. La reazione consiste nell’attacco nucleofilico di una molecola di diazo-reattivo di Ehrlich (cloruro di diazonio) all’atomo di carbonio 9 o 11 del tetrapirrolo con formazione di un primo azodipirrolo e di un prodotto intermedio instabile, che reagisce con una seconda molecola di diazo-reattivo dando luogo ad un secondo azodipirrolo. Per ogni molecola di bilirubina si formano dunque due molecole di azodipirrolo, dosabili spettrofotometricamente. Il diazo-reattivo, che è instabile, deve essere preparato al momento dell’uso facendo reagire il nitrito di sodio con l’acido sulfanilico (Fig. 5.7).

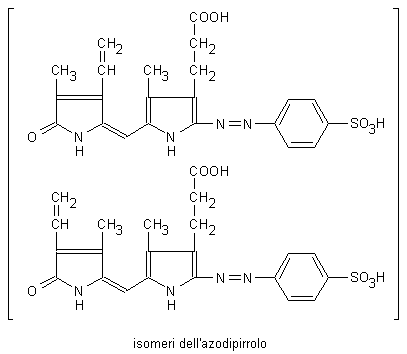
Fig. 5.7. Determinazione della bilirubina con il reattivo di Ehrlich
L’emoglobina interferisce sulla determinazione della bilirubina in modo complesso. Un primo tipo di interferenza si verifica già prima dell’aggiunta del diazo-reattivo ed è dovuto alla distruzione della bilirubina da parte dell’emoglobina; questo inconveniente viene evitato aggiungendo al campione della caffeina, che ha un effetto stabilizzante sulla bilirubina. Un secondo tipo di interferenza si ha nel corso della diazo-reazione per la distruzione della bilirubina da parte della ossiemoglobina e, per ridurre questo effetto, è necessario convertire l’ossiemoglobina in carbossiemoglobina. Una terza causa di interferenza è l’ossidazione della bilirubina da parte dei perossidi che si generano nell’ambiente acido della diazo-reazione a seguito della formazione di un ponte -O-O- tra due molecole di ossiemoglobina con liberazione di eme ferrico; questo effetto è prevenuto aggiungendo agenti riducenti, come ad esempio lo ioduro di potassio. Una quarta interferenza si ha in fase di lettura dell’assorbanza ottica del prodotto della diazo-reazione in quanto vi è una sovrapposizione spettrale fra l’emoglobina e gli azodipirroli in ambiente acido. Inoltre, la formazione di metaemoglobina a partire dall’ossiemoglobina è più rapida nel test che nel bianco e l’interferenza spettrale della metaemoglobina è inferiore rispetto a quella dell’ossiemoglobina. Aggiungendo tartrato alcalino è possibile spostare verso lunghezze d’onda più alte lo spettro di assorbimento degli azo-pigmenti e, inoltre, favorire la formazione di metaemoglobina sia nel test che nel bianco. Tuttavia in queste condizioni si provoca una ulteriore distruzione della bilirubina, che può essere però prevenuta aggiungendo ascorbato prima del tartrato alcalino.
Fin dall’inizio fu visto che la bilirubina presente nelle urine e nel plasma di pazienti con ittero ostruttivo (bilirubina diretta) reagiva direttamente con il diazo-reattivo, mentre la bilirubina nel plasma dei pazienti con ittero emolitico (bilirubina indiretta) richiedeva un agente, come l’alcool etilico, in cui sia la bilirubina che il diazo-reattivo fossero solubili. Successivamente fu stabilito che la frazione indiretta corrispondeva alla bilirubina non coniugata, mentre la frazione diretta era identificabile con la bilirubina glucuronata. Anche la bilirubina glucuronata legata irreversibilmente all’albumina (bilialbumina) reagiva direttamente con il diazo-reattivo, potendo costituire in talune circostanze la maggior parte del pigmento a reazione diretta nel siero di pazienti con ittero colostatico.
Sebbene la distinzione tra bilirubina diretta ed indiretta sia ancora molto utile nella pratica clinica, è ormai assodato che le due frazioni non riflettono accuratamente le concentrazioni di bilirubina non coniugata e coniugata, ma sono influenzate dalle condizioni di reazione (pH, forza ionica, temperatura, esposizione alla luce). Diminuendo il tempo di reazione, è possibile minimizzare la quota di bilirubina non coniugata che reagisce con il diazo-reattivo in assenza di un opportuno solvente, aumentando così la specificità del saggio della bilirubina diretta. Tuttavia, poiché esiste sempre una quota variabile di bilirubina non coniugata che reagisce direttamente con il diazo-reattivo, è impossibile determinare con precisione la quota di bilirubina glucuronata in presenza di una grande quantità di pigmento non coniugato. D’altra parte è noto che la diazo-reazione quantifica solo circa il 70% della bilirubina esterificata.
Le procedure attualmente in uso derivano da quelle proposte da Malloy e Evelyn e da Jendrassik e Grof. Secondo Malloy e Evelyn la reazione deve avvenire a pH 1,2 e la frazione non coniugata deve essere solubilizzata in metanolo. A questo pH l’azo-pigmento ha un massimo di assorbimento a 560 nm. Il metodo di Jendrassik e Grof prevede che la reazione avvenga a pH 6,5 e che l’azo-pigmento sia misurato a 600 nm dopo alcalinizzazione a pH 13. La bilirubina non coniugata è solubilizzata in benzoato di sodio e caffeina. Ulteriori modifiche ai due metodi riguardano il solvente da usare per solubilizzare la bilirubina non coniugata, che può essere il dimetil sulfossido, l’urea o l’antipirina e metanolo.
L’acido sulfanilico può essere sostituito con la p-iodoanilina. Il metodo è molto sensibile e riproducibile, particolarmente in presenza di campioni torbidi, grazie alla possibilità di estrarre gli azo-pigmenti derivati dalla reazione in un solvente organico. Un altro metodo prevede l’uso dell’etil antranilato diazotizzato. Il vantaggio è che la bilirubina non coniugata non reagisce significativamente con questo reattivo a pH 7,2 in un mezzo acquoso, mentre la bilirubina coniugata reagisce completamente.
Un metodo per il dosaggio in fase solida su lastrine a perdere permette di valutare sia la bilirubina totale che le frazioni libera ed esterificata. Nella lastrina per il dosaggio della bilirubina totale, la diazo-reazione avviene in uno strato poroso a pH controllato e gli azo-derivati vengono legati ad un mordente che provoca una modificazione dello spettro d’assorbimento. Per evitare interferenze da parte di altri composti, l’assorbimento viene determinato a due diverse lunghezze d’onda. La lastrina per il dosaggio differenziato della bilirubina non coniugata e di quella esterificata è composta di un primo strato poroso, su cui viene steso il campione di siero e che impedisce la migrazione negli strati sottostanti della bilialbumina, dell’emoglobina e di altre sostanze interferenti. La bilirubina non coniugata e quella esterificata raggiungono invece lo strato sottostante dove si legano ad un mordente che ne modifica lo spettro di assorbimento rendendo differenziabili a diverse lunghezze d’onda le due frazioni. Il metodo è calibrato con bilirubina coniugata alla taurina, che costituisce, in queste condizioni, un surrogato accettabile della bilirubina glucuronata.
5.2.1c. Purificazione dell’analita
Una ulteriore procedura si basa su una reazione di trans-esterificazione in metanolo alcalino dei pigmenti che consente la conversione quantitativa dei mono- e diglucuronilderivati della bilirubina nei corrispondenti mono- e dimetilesteri. I prodotti della reazione e la bilirubina non coniugata, che non viene modificata, sono estratti in metanolo e analizzati mediante cromatografia su strato sottile o mediante cromatografia liquida ad alta pressione (HPLC). La prima tecnica permette di ottenere solo informazioni qualitative o semi-quantitative, mentre l’HPLC è in grado, con l’ausilio di uno standard interno, di quantificare accuratamente le varie frazioni di pigmento.
5.2.1d. Bilirubina libera e capacità legante residua dell’albumina
La bilirubina non glucuronata libera può essere separata dalla frazione legata all’albumina mediante tecniche cromatografiche di gel-filtrazione e quindi dosata mediante una diazo-reazione. In alternativa si può ossidare la bilirubina libera a biliverdina incolore mediante una ossidasi o una perossidasi specifica, avendo l’accortezza di misurare la velocità iniziale della reazione per non spostare significativamente della nuova bilirubina dal complesso bilirubina-albumina. La bilirubina legata all’albumina può essere dosata sfruttando il fatto che questa, a differenza della forma libera, è fluorescente. La capacità legante residua dell’albumina può essere misurata titolando l’albumina con bilirubina o con uno dei numerosi coloranti che si legano al sito primario o secondario dell’albumina per la bilirubina.
5.2.2. Preparazione del campione ed intervalli di riferimento
La bilirubina totale può essere dosata mediante la diazo-reazione sia nel plasma che nel siero. Il siero è da preferirsi quando si usa il metodo di Malloy e Evelyn in quanto l’aggiunta di alcool può far precipitare le proteine del plasma e falsare l’analisi. La bilirubina coniugata può essere dosata indifferentemente nel plasma o nel siero, così come nelle urine. I campioni di liquor possono essere utilizzati per il dosaggio sia della bilirubina totale che della frazione diretta. I campioni con emolisi anche lieve devono essere scartati. Poiché la bilirubina può essere distrutta alla luce e al calore, è necessario eseguire l’analisi il prima possibile senza esporre il campione a fonti luminose. Per il dosaggio della bilirubina libera sono necessari almeno 0,1 mL di siero; altri 0,5 mL sono necessari per il dosaggio della capacità legante residua dell’albumina.
La concentrazione di bilirubina totale nel siero è di norma inferiore a 15 mg/L (25.7 μmol/L). In entrambi i sessi la bilirubinemia tende ad aumentare con la pubertà e diminuisce dopo la terza decade. Dopo la pubertà la bilirubinemia è generalmente più elevata nei maschi che nelle femmine. La concentrazione della bilirubina coniugata è inferiore a 2 mg/L.
L’albumina deve essere in un eccesso molare rispetto la bilirubina non coniugata per impedire l’insorgenza di effetti tossici. Si considera pertanto che concentrazioni di bilirubina non coniugata nel siero inferiori a 200 mg/L siano sotto il livello di guardia. Se la capacità legante residua dell’albumina è inferiore a 50 mg/L il rischio di kernittero è consistente. Quando la bilirubina libera, ossia la quota di bilirubina non coniugata con l’acido glucuronico e non veicolata all’albumina, supera i 12 m g/L (20 nmol/L) è necessario intervenire con una exanguinotrasfusione.
| aggiornamento: 19/07/11 |