|
|
Costantino Salerno |
|
|
Appunti di Biochimica Clinica |
|
|
1. INTRODUZIONE
Il termine "biochimica clinica" si può riferire allo stesso tempo a due diversi aspetti dell’attività biomedica: (1) allo studio (in continua evoluzione) dei meccanismi biochimici alla base dei processi fisiopatologici; (2) al lavoro del medico volto alla determinazione di singoli analiti e all’utilizzo dei dati così ottenuti a fini diagnostici.
Dal primo punto di vista, la biochimica clinica è un settore della ricerca le cui acquisizioni possono trovare larga e feconda applicazione nel campo sanitario. Non soltanto le malattie metaboliche congenite o acquisite, ma tutte le forme morbose sono sostenute o accompagnate da disturbi biochimici. Rapporti molto stretti ed evidenti esistono inoltre tra metabolismo e farmaci, come risulta dallo studio degli antimetaboliti e dallo sviluppo della chemioterapia, o tra carenze vitaminiche e lesioni biochimiche. D’altra parte, il notevole sviluppo che la medicina ha dato alla biochimica può essere, almeno in parte, dovuto alle positive ricadute che la ricerca biochimica di base ha sullo sviluppo delle conoscenze e sui possibili interventi in campo clinico.
Dal secondo punto di vista, la biochimica clinica è un’attività razionale che consiste nel sapersi avvalere di opportune tecniche analitiche per acquisire informazioni e prendere decisioni in condizione di incertezza. Dal confronto fra ciò che osserva e ciò che sa, il medico individua nel singolo caso concreto quale potrebbe essere la migliore strada da seguire fra le possibili alternative e ne illustra i pro e i contro al paziente, al quale spetta la decisione finale.
La biochimica clinica intesa come scienza si differenzia dalla biochimica clinica intesa come parte di un processo diagnostico per la diversità degli scopi e dei metodi. Mentre nella ricerca di base l’ottenimento del dato analitico è parte del processo di verifica dell’ipotesi di lavoro ed ha una validità indipendente dal risultato, la richiesta di un test diagnostico è giustificata solo da una sua effettiva utilità nell’indirizzare future decisioni nell’interesse del paziente. Il clinico deve essere al corrente di tutte le indagini di cui può disporre, ma deve altresì essere in grado di valutarne i vantaggi e i limiti.
Va comunque ricordato che il laboratorio di analisi si è via via trasformato da semplice ausilio per la conferma della diagnosi in efficace strumento per l’approfondimento e l’estensione dell’esame clinico, nonché, attraverso la raccolta di un numero sempre più elevato di dati all’ingresso del malato in ospedale, per la preparazione al ragionamento diagnostico. Prescrivere un test di laboratorio è facile e non vi è dubbio che spesso i test sono richiesti senza porre la dovuta attenzione al loro reale valore diagnostico in una specifica situazione clinica. I laboratoristi cercano di scoraggiare questo atteggiamento, ma non li favorisce certo l’uso che essi per primi fanno dei termini "test di routine" (riferito ai test di più comune impiego) oppure "laboratori di routine" (riferito ai servizi dove questi test sono eseguiti).
Il cattivo uso del laboratorio a fini diagnostici ha,
peraltro, effetti economici significativamente negativi per l’alto
costo delle procedure analitiche, cosa che porta ad una non
razionale gestione della struttura sanitaria e ad un danno per il
paziente che ne è nel contempo finanziatore ed utilizzatore. Le
analisi cliniche sono uno dei capitoli che meno risente delle
misure di riduzione della spesa. In Italia, malgrado l’introduzione
di ticket sanitari, l’esborso complessivo per le
analisi in ambiente ospedaliero e convenzionato esterno è
rimasto pressoché stabile
![]() . La ragione forse sta nel
fatto che finora si è agito soltanto sul paziente con misure
punitive, anziché sul medico che in fin dei conti è il solo a
poter autorizzare le indagini cliniche. Se si esamina, infatti,
con che frequenza sono richiesti gli esami di laboratorio nei
diversi paesi europei, si può osservare che in media il medico
di base prescrive almeno un esame di laboratorio al 7,7% dei
pazienti che si presentano a lui per una visita. Questo valore
varia tuttavia notevolmente da paese a paese: i medici svizzeri
sono nettamente in testa nella classifica con il 15,5% mentre
quelli britannici sono i più parsimoniosi con il 5,1% (vedi
Fig. 1.1). E’ interessante notare che
spesso il medico non è selettivo nella richiesta, tendendo cioè
ad indagare su diversi disturbi nel corso della stessa visita: il
Belgio è al primo posto per quanto riguarda questo punto con 4,1
gruppi di esami per paziente, seguito dall’Italia, la Spagna
e il Portogallo, mentre i medici britannici sono al gradino più
basso della graduatoria con una media di 1,6 gruppi di esami per
paziente. Per quanto riguarda il tipo di esame, l’ematocrito
e l’emocromo (21,2% di tutti i test; vedi
Par. 7.1) sono i più
richiesti, seguiti dalla glicemia (17,4%; vedi
Par. 2.1). Purtroppo c’è
ragione per ritenere che diversi fattori di carattere non
strettamente sanitario possano influire sulla decisione del
medico di chiedere più o meno analisi. Le variabili che
influenzano maggiormente la prescrizione di esami sono infatti il
numero di medici di base per mille abitanti (la prescrizione
aumenta con la densità dei medici) e il numero di visite per
settimana (il medico prescrive meno esami se vede tanti pazienti),
suggerendo che motivi di competizione o la mancanza di tempo
disponibile possano influenzare il comportamento del medico. Un
altro fattore molto importante che condiziona la frequenza di
richieste di analisi di laboratorio è il tipo di organizzazione
sanitaria che è stata adottata: nei paesi dove c’è un
pagamento del medico a prestazione vengono infatti prescritti
molti più esami che nei paesi dove invece questi rientrano nella
quota capitaria.
. La ragione forse sta nel
fatto che finora si è agito soltanto sul paziente con misure
punitive, anziché sul medico che in fin dei conti è il solo a
poter autorizzare le indagini cliniche. Se si esamina, infatti,
con che frequenza sono richiesti gli esami di laboratorio nei
diversi paesi europei, si può osservare che in media il medico
di base prescrive almeno un esame di laboratorio al 7,7% dei
pazienti che si presentano a lui per una visita. Questo valore
varia tuttavia notevolmente da paese a paese: i medici svizzeri
sono nettamente in testa nella classifica con il 15,5% mentre
quelli britannici sono i più parsimoniosi con il 5,1% (vedi
Fig. 1.1). E’ interessante notare che
spesso il medico non è selettivo nella richiesta, tendendo cioè
ad indagare su diversi disturbi nel corso della stessa visita: il
Belgio è al primo posto per quanto riguarda questo punto con 4,1
gruppi di esami per paziente, seguito dall’Italia, la Spagna
e il Portogallo, mentre i medici britannici sono al gradino più
basso della graduatoria con una media di 1,6 gruppi di esami per
paziente. Per quanto riguarda il tipo di esame, l’ematocrito
e l’emocromo (21,2% di tutti i test; vedi
Par. 7.1) sono i più
richiesti, seguiti dalla glicemia (17,4%; vedi
Par. 2.1). Purtroppo c’è
ragione per ritenere che diversi fattori di carattere non
strettamente sanitario possano influire sulla decisione del
medico di chiedere più o meno analisi. Le variabili che
influenzano maggiormente la prescrizione di esami sono infatti il
numero di medici di base per mille abitanti (la prescrizione
aumenta con la densità dei medici) e il numero di visite per
settimana (il medico prescrive meno esami se vede tanti pazienti),
suggerendo che motivi di competizione o la mancanza di tempo
disponibile possano influenzare il comportamento del medico. Un
altro fattore molto importante che condiziona la frequenza di
richieste di analisi di laboratorio è il tipo di organizzazione
sanitaria che è stata adottata: nei paesi dove c’è un
pagamento del medico a prestazione vengono infatti prescritti
molti più esami che nei paesi dove invece questi rientrano nella
quota capitaria.
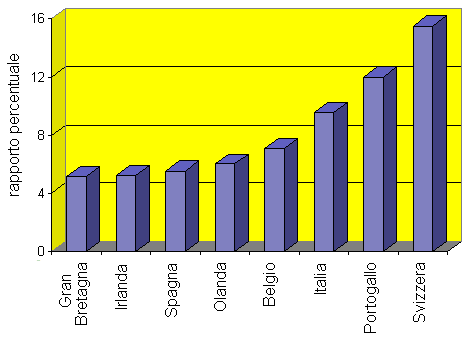
Fig. 1.1. Frequenza di richiesta di esami di laboratorio in alcuni paesi europei (1995). L’istogramma mostra la percentuale dei pazienti a cui viene prescritto almeno un esame di laboratorio a seguito di una visita presso un ambulatorio di medicina si base.
1.1. TEORIA DELLE DECISIONI E METODO BAYESIANO
I metodi che si possono seguire per risolvere un quesito diagnostico sono diversi e il loro studio rientra in quello più generale riguardante la teoria delle decisioni. Una strategia comunemente usata per valutare la bontà di un test è quella di determinare il suo valore predittivo, ossia di stabilire quale probabilità ha un soggetto positivo al test di essere effettivamente affetto dalla malattia in esame. Per fare ciò è necessario conoscere la prevalenza della malattia nella popolazione in esame, nonché la sensibilità e la specificità del test. Presupposto essenziale per il calcolo della sensibilità e della specificità del test è che la presenza o l’assenza della malattia possano essere stabilite con certezza mediante un qualche metodo incontrovertibile: in alcuni casi si può fare riferimento al risultato dell’esame istologico o autoptico, in altri casi ci si può basare sull’esito clinico o su qualche altro evento conclusivo più o meno definitivo. Si tratta cioè di calcolare, sulla base delle frequenze relative sperimentalmente osservabili, la probabilità condizionata, p(x/ξ), della risposta x all’analisi quando è presente un evento morboso ξ. Così facendo, infatti, se in un nuovo gruppo di pazienti è nota solo la probabilità a priori p(ξ) dell’evento ξ, è possibile operare una inferenza statistica (teorema di Bayes) che ci porta a determinare la probabilità condizionata p(ξ/x). Quest’ultima rappresenta la probabilità a posteriori dell’evento ξ una volta che è nota la risposta x all’analisi e ne è perciò presumibilmente una stima migliore.
Il caso più semplice si ha quando è possibile confrontare
fra loro due sole ipotesi (presenza della malattia, ξ1; assenza della malattia, ξ0) e quando l’analisi dà
luogo solo a due risposte alternative (positiva, x1;
negativa, x0). Un esempio è dato dal test
di agglutinazione al lattice per i fattori reumatoidi in una
sospetta artrite reumatoide
![]() : le due ipotesi a confronto
sono l’essere affetti da artrite reumatoide (ξ1) o il non avere questa
malattia (ξ0); le due
risposte al test sono la presenza (x1) o
l’assenza (x0) di agglutinazione sul
vetrino. Come mostrato in Tab.
1.I, la popolazione esaminata può essere divisa
in 4 gruppi: (1) soggetti con artrite reumatoide e test
positivo (veri positivi); (2) soggetti con artrite
reumatoide e test negativo (falsi negativi); (3)
soggetti non affetti da artrite reumatoide con test
positivo (falsi positivi); (4) soggetti non affetti da
artrite reumatoide con test negativo (veri negativi).
: le due ipotesi a confronto
sono l’essere affetti da artrite reumatoide (ξ1) o il non avere questa
malattia (ξ0); le due
risposte al test sono la presenza (x1) o
l’assenza (x0) di agglutinazione sul
vetrino. Come mostrato in Tab.
1.I, la popolazione esaminata può essere divisa
in 4 gruppi: (1) soggetti con artrite reumatoide e test
positivo (veri positivi); (2) soggetti con artrite
reumatoide e test negativo (falsi negativi); (3)
soggetti non affetti da artrite reumatoide con test
positivo (falsi positivi); (4) soggetti non affetti da
artrite reumatoide con test negativo (veri negativi).
Tab. 1.I - Test di agglutinazione al lattice per i fattori reumatoidi in ambiente specialistico
Artrite Reumatoide |
||
presente |
assente | |
| Test positivo | veri positivi: 1395 |
falsi positivi: 713 |
| Test negativo | falsi negativi: 475 |
veri negativi: 2142 |
Totali |
malati: 1870 |
non malati: 2855 |
| Probabilità a priori p(ξ1) | ||
| prevalenza | = 1870 / (1870 + 2855 ) = 0,396 | |
| Probabilità condizionate p(x1/ξ1) e p(x0/ξ0) | ||
| sensibilità | = 1395 / 1870 = 0,746 | |
| specificità | = 2142 / 2855 = 0,750 | |
| Probabilità a posteriori p(ξ1/x1) | ||
| valore predittivo | = 1395 / (1395 + 713) = 0,662 | |
Le probabilità condizionate p(x1/ξ1) e p(x0/ξ0) rappresentano,
rispettivamente, la sensibilità (rapporto fra veri positivi e
tutti i malati) e la specificità (rapporto fra veri negativi e
tutti i soggetti non malati) del test. La probabilità a
priori p(ξ1) rappresenta la
prevalenza della malattia nel gruppo dei soggetti esaminati. La
probabilità condizionata a posteriori p(ξ1/x1)
rappresenta il valore predittivo del test (rapporto fra
veri positivi e tutti i positivi
![]() ). E’ ovvio che il test
sarà utile se e solo se la probabilità a posteriori (valore
predittivo) sarà diversa da quella a priori (prevalenza),
ossia se il medico avrà maggiore probabilità di formulare una
diagnosi corretta (affermando che il paziente è effettivamente
affetto dalla malattia in esame o escludendo tale ipotesi) dopo aver ricevuto il responso
dell’analisi rispetto a prima. Quando la composizione della
popolazione in esame non è nota altrimenti, il valore predittivo
può essere calcolato mediante l’equazione:
). E’ ovvio che il test
sarà utile se e solo se la probabilità a posteriori (valore
predittivo) sarà diversa da quella a priori (prevalenza),
ossia se il medico avrà maggiore probabilità di formulare una
diagnosi corretta (affermando che il paziente è effettivamente
affetto dalla malattia in esame o escludendo tale ipotesi) dopo aver ricevuto il responso
dell’analisi rispetto a prima. Quando la composizione della
popolazione in esame non è nota altrimenti, il valore predittivo
può essere calcolato mediante l’equazione:
| valore predittivo = prevalenza x . |
sensibilità
|
|
prevalenza x sensibilità + (1 - prevalenza) x (1 -
specificità)
|
Si può facilmente osservare che il test è inutile (valore
predittivo = prevalenza) quando la percentuale dei risultati
positivi nel gruppo dei soggetti malati è uguale a quella nel
gruppo dei soggetti non affetti dalla malattia in esame (sensibilità = 1 - specificità) o
quando si ha già la certezza dell’assenza (prevalenza = 0)
o della presenza (prevalenza = 1) della malattia. Negli altri
casi il valore predittivo si discosta dalla prevalenza via via che aumenta
quest’ultimo parametro (Tab. 1.II).
Da ciò consegue che la fiducia che possiamo accordare al
risultato di un test è minore se la malattia da
diagnosticare è rara nel gruppo di pazienti da noi osservato. Ciò
può capitare, ad esempio, quando operiamo nell’ambito della
medicina di base dove, di norma, una patologia di interesse
settoriale è osservabile con minore frequenza rispetto ad un
ambiente specialistico
![]() . A tale proposito è interessante notare
che, benché il test di Guthrie per lo screening della fenilchetonuria (vedi Par.
14.6.4a) abbia sensibilità e specificità prossime al
100%, il suo valore predittivo è solo del 10% perché la
prevalenza della malattia è estremamente bassa (circa 1:12.000).
Inoltre, a corollario di quanto detto, vi è l’avvertenza
che un test biochimico, specie se costoso o invasivo, non
dovrebbe essere mai eseguito alla cieca, senza aver sottoposto il
paziente a quella serie di esami, propri della semeiotica fisica,
necessari per restringere preliminarmente le possibilità
diagnostiche.
. A tale proposito è interessante notare
che, benché il test di Guthrie per lo screening della fenilchetonuria (vedi Par.
14.6.4a) abbia sensibilità e specificità prossime al
100%, il suo valore predittivo è solo del 10% perché la
prevalenza della malattia è estremamente bassa (circa 1:12.000).
Inoltre, a corollario di quanto detto, vi è l’avvertenza
che un test biochimico, specie se costoso o invasivo, non
dovrebbe essere mai eseguito alla cieca, senza aver sottoposto il
paziente a quella serie di esami, propri della semeiotica fisica,
necessari per restringere preliminarmente le possibilità
diagnostiche.
Tab. 1.II. Confronto fra popolazioni con diversa prevalenza di malattia. I risultati sono stati ottenuti applicando un medesimo test con sensibilità e specificità del 90% a due ipotetiche popolazioni di 100 individui ciascuna, con una prevalenza della malattia in esame rispettivamente del 10% e del 50%.
Prevalenza della malattia = 10% |
Prevalenza della malattia = 50% |
||||||||
TEST |
malati |
non malati |
TEST |
malati |
non malati |
||||
| positivo | 9 |
9 |
18 |
positivo | 45 |
5 |
50 |
||
| negativo | 1 |
81 |
82 |
negativo | 5 |
45 |
50 |
||
10 |
90 |
100 |
50 |
50 |
100 |
||||
valore predittivo del test = 50% |
valore predittivo del test = 90% |
||||||||
1.1.1. Test utilizzanti grandezze continue
Quando il risultato dell’analisi biochimica dà luogo ad
una grandezza continua (ad esempio quando è espresso in termini
di concentrazione dell’analita) è ugualmente possibile
utilizzare il metodo descritto precedentemente per i test del
tipo "tutto-o-nulla", a patto che si scelga un
opportuno valore discriminante (cut-off point) che
permetta di separare i risultati positivi da quelli negativi. Un
esempio è offerto dalla determinazione della colesterolemia in
soggetti di controllo ed in pazienti eterozigoti per l’ipercolesterolemia
familiare
![]() (Fig. 1.2). I livelli di
colesterolo plasmatico si distribuiscono nelle due popolazioni su
due ampi intervalli di concentrazione in parte sovrapposti. E’
evidente che la scelta del valore discriminante influenzerà la
sensibilità e la specificità e, conseguentemente, il valore
predittivo del test in esame. Se vengono scelti valori di cut-off
molto elevati (per esempio, superiori a 3 g/L) si potrà evitare
di avere dei risultati falsi positivi, ma il gruppo di pazienti
posti a sinistra rispetto al cut-off sulla curva di
distribuzione di frequenza sarà erroneamente considerato
negativo al test. Se vengono scelti valori di cut-off
molto bassi (per esempio, inferiori a 1 g/L) la sensibilità del test
diverrà molto elevata, ma gran parte dei soggetti di
controllo risulterà falsamente positiva all’analisi.
Chiaramente un buon test deve essere contemporaneamente
sensibile e specifico, ossia, in altri termini, deve dare
risultati positivi nei soggetti malati e risultati negativi nei
soggetti di controllo. Nel caso in esame, perciò, il valore di cut-off
dovrà essere scelto opportunamente nell’intervallo
compreso tra 1 g/L e 3 g/L. La strategia da seguire varierà a
seconda che sia nota o meno la funzione di perdita da minimizzare
(Fig. 1.2). I livelli di
colesterolo plasmatico si distribuiscono nelle due popolazioni su
due ampi intervalli di concentrazione in parte sovrapposti. E’
evidente che la scelta del valore discriminante influenzerà la
sensibilità e la specificità e, conseguentemente, il valore
predittivo del test in esame. Se vengono scelti valori di cut-off
molto elevati (per esempio, superiori a 3 g/L) si potrà evitare
di avere dei risultati falsi positivi, ma il gruppo di pazienti
posti a sinistra rispetto al cut-off sulla curva di
distribuzione di frequenza sarà erroneamente considerato
negativo al test. Se vengono scelti valori di cut-off
molto bassi (per esempio, inferiori a 1 g/L) la sensibilità del test
diverrà molto elevata, ma gran parte dei soggetti di
controllo risulterà falsamente positiva all’analisi.
Chiaramente un buon test deve essere contemporaneamente
sensibile e specifico, ossia, in altri termini, deve dare
risultati positivi nei soggetti malati e risultati negativi nei
soggetti di controllo. Nel caso in esame, perciò, il valore di cut-off
dovrà essere scelto opportunamente nell’intervallo
compreso tra 1 g/L e 3 g/L. La strategia da seguire varierà a
seconda che sia nota o meno la funzione di perdita da minimizzare
![]() .
.
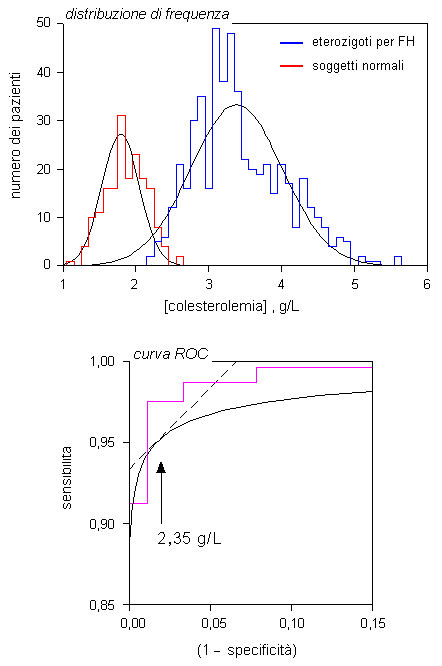
Fig. 1.2. Distribuzione dei livelli di colesterolo sierico in un gruppo di soggetti normali ed in un gruppo di eterozigoti per l’ipercolesterolemia familiare (FH) in uno studio comparativo sulla popolazione giapponese (1986). Le distribuzioni di frequenza relative alle due popolazioni danno luogo a curve in parte sovrapposte con un andamento grossolanamente gaussiano. Il valore discriminante che minimizza la frequenza degli errori diagnostici è stato determinato con l’ausilio di un grafico (curva ROC) che mette in relazione la sensibilità con il complemento ad uno della specificità al test, calcolati per una serie decrescente di valori di cut-off. Il grafico è stato calcolato sulla base sia dei dati sperimentali (linea spezzata in colore) che delle guassiane interpolanti (curva in nero). Il valore discriminate prescelto (2,35 g/L) è quello che corrisponde al punto della curva ROC tangente ad una retta con coefficente angolare unitario (linea tratteggiata).
Quando la scelta più opportuna del valore di cut-off
è problematica, può risultare utile l’impiego di una curva
ROC
![]() . Questa viene costruita calcolando la sensibilità
e la specificità del test ad ogni singolo valore di cut-off
e ponendo in grafico la sensibilità in funzione del complemento
ad uno della specificità (1 - specificità). In tal modo il
valore delle ascisse nel grafico sarà proporzionale al numero
dei falsi positivi, mentre il valore delle ordinate sarà
proporzionale al numero dei veri positivi al test. Come
mostrato in Fig. 1.3, la curva ROC deve
passare per i punti (0, 0) e (1, 1) ed è definita nell’intervallo
. Questa viene costruita calcolando la sensibilità
e la specificità del test ad ogni singolo valore di cut-off
e ponendo in grafico la sensibilità in funzione del complemento
ad uno della specificità (1 - specificità). In tal modo il
valore delle ascisse nel grafico sarà proporzionale al numero
dei falsi positivi, mentre il valore delle ordinate sarà
proporzionale al numero dei veri positivi al test. Come
mostrato in Fig. 1.3, la curva ROC deve
passare per i punti (0, 0) e (1, 1) ed è definita nell’intervallo
0 ≤ (1 - specificità) ≤ 1
L’area sottesa dalla curva nell’intervallo di definizione equivale alla probabilità, per un soggetto scelto a caso fra quelli affetti da malattia, di avere un risultato al test diverso da quello ottenuto da un soggetto scelto a caso nella popolazione di controllo. Quando non vi è differenza fra le due popolazioni riguardo alla variabile in esame, la curva ROC si riduce ad una retta con coefficiente angolare unitario e l’area sottesa è uguale ad 1/2. Quando vi è una perfetta separazione fra i due gruppi (ossia, quando non vi è alcuna sovrapposizione fra le due distribuzioni di frequenza) l’area sottesa è uguale ad 1 e la curva si schiaccia sull’asse delle ordinate raggiungendo in alto a sinistra il punto con coordinate (0, 1).
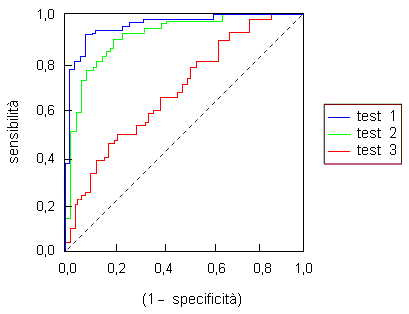
Fig. 1.3. Curve ROC per tre ipotetici test. Le distribuzioni di frequenza in esame sono maggiormente separate fra loro nel test 1 rispetto al test 2 e nel test 2 rispetto al test 3. La linea punteggiata si riferisce al caso in cui non vi sia alcuna differenza fra le due popolazioni rispetto alla variabile in esame.
Se il quadro morboso è caratterizzato da un aumento della concentrazione dell’analita in esame, la diminuzione del valore di cut-off determinerà, in un primo momento, un incremento della quota dei veri positivi (e quindi un incremento della sensibilità del metodo, come è evidenziato dai punti sulla branca ascendente della curva ROC). In un secondo momento, l’incremento della sensibilità del metodo si farà meno pronunciato, mentre la specificità dell’analisi sarà via via ridotta con l’inclusione di un numero sempre maggiore di soggetti di controllo fra i positivi al test. Quando ciò accade (ossia per un valore della tangente alla curva ROC inferiore all’unità) non è più conveniente diminuire ulteriormente il valore di cut-off.
1.2. VARIABILITA’ BIOLOGICA
La variabilità dei parametri biochimici è influenzata da numerosi fattori inerenti al paziente. Questi fattori possono essere distinti in endogeni (età, sesso, massa corporea) ed in esogeni (variazioni cronobiologiche, stress, attività fisica, alimentazione, postura).
Ad esempio, l’attività della fosfatasi alcalina plasmatica (vedi Par. 9.1.4) è più elevata nei bambini che negli adulti, in particolare durante l’accelerato accrescimento puberale. In generale, le differenze delle variabili biochimiche che dipendono dal sesso sono meno pronunciate prima della pubertà, mentre nell’adulto tendono a ridursi dopo la menopausa. A parte le più note differenze di concentrazione ormonale plasmatica, i due sessi possono presentare delle variazioni rispetto anche altri analiti, quando il loro metabolismo è influenzato dagli ormoni sessuali. Così la concentrazione del colesterolo plasmatico (vedi Par. 3.3) tende ad essere più elevata nel maschio che nella femmina prima della menopausa, mentre dopo questo evento i valori della donna tendono ad aumentare. Per quanto riguarda la massa corporea, si può ricordare che la concentrazione dell’insulina plasmatica e dei trigliceridi tende ad essere più elevata nei soggetti obesi (vedi Par. 3.5) . La creatinina, che è normalmente prodotta dal metabolismo del tessuto muscolare scheletrico è, a parità di filtrazione glomerulare, più concentrata nel plasma di un soggetto con maggiore sviluppo della muscolatura (vedi Par. 6.4).
La concentrazione del 25-idrossicolecalciferolo varia con andamento stagionale con valori più elevati d’estate (nell’emisfero settentrionale) che d’inverno (vedi Par. 10.9d). Le concentrazioni delle gonadotropine, degli estrogeni e del progesterone variano nel corso del ciclo mestruale nella donna in età fertile (vedi Par. 10.6.1). Il cortisolo è caratterizzato da un ritmo diurno con un minimo intorno alla mezzanotte ed un massimo alle 8-9 del mattino (vedi Par. 10.5.3). Gli ormoni ipofisari e surrenalici (ACTH, cortisolo, prolattina, ormone della crescita, catecolamine plasmatiche) aumentano tutti in risposta allo stress. Per quanto riguarda l’attività fisica, questa può determinare, se particolarmente intensa, un aumento dell’attività plasmatica della creatina chinasi (vedi Par. 9.1.2). L’assunzione di alimenti determina un aumento della concentrazione plasmatica di glucosio, trigliceridi ed insulina, mentre un pasto particolarmente ricco di proteine è seguito da un aumento della sintesi e della concentrazione plasmatica di urea (vedi Par. 6). Molti parametri sono infine influenzati dalla postura. L’esempio più noto è rappresentato dalla renina e dall’aldosterone, le cui concentrazioni plasmatiche sono più elevate in posizione eretta rispetto a quella supina per variazioni nel flusso ematico renale (vedi Par. 10.5.3).
1.2.1. Valori di riferimento
Nell’osservare un dato analitico, non è infrequente
porsi il quesito se esso sia normale
![]() . Per facilitare l’interpretazione
dei risultati, il valore ottenuto dall’analisi è perciò
generalmente messo a confronto con dei "valori di
riferimento", stabiliti in base a quanto precedentemente
ottenuto con soggetti (soggetti di riferimento) accuratamente
selezionati secondo criteri predefiniti, come l’età, il
sesso, la condizione di salute, etc. Di consueto ed al fine di
escludere soggetti devianti a causa di una qualche patologia non
altrimenti rilevabile, i limiti dell’intervallo di
riferimento sono stabiliti in modo da comprendere solo il 95% del
campione con esclusione delle frange estreme dei valori,
comprendendo cioè i soggetti dal percentile 2,5 al percentile 97,5
o, nel caso di una distribuzione gaussiana
. Per facilitare l’interpretazione
dei risultati, il valore ottenuto dall’analisi è perciò
generalmente messo a confronto con dei "valori di
riferimento", stabiliti in base a quanto precedentemente
ottenuto con soggetti (soggetti di riferimento) accuratamente
selezionati secondo criteri predefiniti, come l’età, il
sesso, la condizione di salute, etc. Di consueto ed al fine di
escludere soggetti devianti a causa di una qualche patologia non
altrimenti rilevabile, i limiti dell’intervallo di
riferimento sono stabiliti in modo da comprendere solo il 95% del
campione con esclusione delle frange estreme dei valori,
comprendendo cioè i soggetti dal percentile 2,5 al percentile 97,5
o, nel caso di una distribuzione gaussiana
![]() ,
i soggetti con valori dell’analita distanti di non più di 2
deviazioni standard dalla media. Ovvie considerazioni statistiche
fanno prevedere di trovare 5 risultati fuori dall’intervallo
di riferimento ogni 100 soggetti esaminati. E’ altresì
ovvio che ogni singolo paziente ha una probabilità maggiore di
ottenere un dato fuori dal riferimento quando è sottoposto ad un
elevato numero di analisi non correlate fra loro. Questo può
capitare, ad esempio, in occasione di un check-up, quando
si vuole indagare su un ampio spettro di possibili alterazioni.
In tal caso diventa molto probabile che almeno una delle risposte
ai test risulti fuori intervallo senza che per questo
debba essere considerata un indizio di malattia (Tab. 1.III).
,
i soggetti con valori dell’analita distanti di non più di 2
deviazioni standard dalla media. Ovvie considerazioni statistiche
fanno prevedere di trovare 5 risultati fuori dall’intervallo
di riferimento ogni 100 soggetti esaminati. E’ altresì
ovvio che ogni singolo paziente ha una probabilità maggiore di
ottenere un dato fuori dal riferimento quando è sottoposto ad un
elevato numero di analisi non correlate fra loro. Questo può
capitare, ad esempio, in occasione di un check-up, quando
si vuole indagare su un ampio spettro di possibili alterazioni.
In tal caso diventa molto probabile che almeno una delle risposte
ai test risulti fuori intervallo senza che per questo
debba essere considerata un indizio di malattia (Tab. 1.III).
Tab. 1.III. Probabilità di avere una risposta fuori l’intervallo di riferimento per un paziente sottoposto ad n analisi. La tabella è stata costruita a solo scopo didattico nell’ipotesi che le analisi non siano correlate fra loro, cioè che il risultato positivo ad una prima analisi non aumenti la probabilità di ottenere un risultato positivo ad una analisi successiva.
Numero di analisi eseguite |
probabilità |
1 |
0,05 |
2 |
0,10 |
3 |
0,14 |
5 |
0,23 |
10 |
0,40 |
20 |
0,64 |
50 |
0,92 |
1.3. ERRORI DI MISURA
Col termine "errore di misura" si definisce la
differenza fra il valore ottenuto e il valore vero. Questa
differenza è funzione di numerose variabili. Una prima
distinzione viene usualmente fatta fra "errori grossolani"
(o sbagli) ed "errori statistici propriamente detti"
![]() .
Gli errori grossolani sono in gran parte eliminabili mediante una
pianificazione oculata e una verifica dei passaggi critici e
possono scaturire, ad esempio, dall’aggiunta di un reagente
sbagliato, dall’aver rovesciato una parte del contenuto
della provetta, da un calcolo scorretto o da una trascrizione
errata. Gli errori statistici propriamente detti non sono invece
mai eliminabili del tutto, anche se è possibile avere una loro
stima, ammesso che sia costruibile un’opportuna funzione di
probabilità determinando la frequenza con cui vengono ottenuti i
diversi valori relativi alla variabile in esame. Nelle analisi
biochimiche questa funzione approssima a volte, ma non
necessariamente, una curva gaussiana. Quando ciò avviene, i
limiti ±2s
o ± 3s imposti alla media dei dati
assicurano che esiste meno di una probabilità su 20 o,
rispettivamente, su 300 che il valore cercato cada al di fuori
dell’intervallo considerato.
.
Gli errori grossolani sono in gran parte eliminabili mediante una
pianificazione oculata e una verifica dei passaggi critici e
possono scaturire, ad esempio, dall’aggiunta di un reagente
sbagliato, dall’aver rovesciato una parte del contenuto
della provetta, da un calcolo scorretto o da una trascrizione
errata. Gli errori statistici propriamente detti non sono invece
mai eliminabili del tutto, anche se è possibile avere una loro
stima, ammesso che sia costruibile un’opportuna funzione di
probabilità determinando la frequenza con cui vengono ottenuti i
diversi valori relativi alla variabile in esame. Nelle analisi
biochimiche questa funzione approssima a volte, ma non
necessariamente, una curva gaussiana. Quando ciò avviene, i
limiti ±2s
o ± 3s imposti alla media dei dati
assicurano che esiste meno di una probabilità su 20 o,
rispettivamente, su 300 che il valore cercato cada al di fuori
dell’intervallo considerato.
Gli errori e gli sbagli possono a loro volta essere divisi in due gruppi: quelli che dipendono dall’analisi stessa e quelli che prescindono da essa, ma riguardano il trattamento preanalitico del campione biologico (prelievo, spedizione, conservazione, etc.) o la refertazione (schedatura, trascrizione del dato, etc.). Infine, a seconda che l’errore sia imputabile ad una eccessiva dispersione dei dati o ad una insufficiente stima del valore medio, si parlerà rispettivamente di scarsa precisione o scarsa accuratezza dell’analisi. La variazione nel tempo di questi parametri influenzerà, a sua volta, l’attendibilità del dato analitico.
1.3.1. Precisione
La precisione si riferisce alla variazione dei risultati
ottenuti con un particolare metodo quando l’analisi viene
eseguita ripetutamente sullo stesso campione
![]() .
Essa include due elementi: la ripetibilità (o precisione entro
la serie) e la riproducibilità (o precisione fra le serie).
.
Essa include due elementi: la ripetibilità (o precisione entro
la serie) e la riproducibilità (o precisione fra le serie).
La ripetibilità è intesa come la deviazione fra le misure
eseguite da un singolo operatore, non attribuibile a
significative variazioni della risposta dell’apparecchiatura
nel tempo. Essa può essere stabilita eseguendo in parallelo un
congruo numero di determinazioni (almeno 30) sul medesimo
campione. Un modo operativamente più semplice per valutare la
ripetibilità delle analisi durante un lavoro di routine
è quello di eseguire in doppio le analisi di almeno 15 campioni,
avendo cura di distribuire a caso i duplicati entro la stessa
serie di analisi e di utilizzare campioni che diano luogo a
risultati tutti distribuiti in un ambito ragionevolmente
ristretto
![]() .
.
La riproducibilità è intesa come la deviazione fra le misure eseguite in tempi diversi non necessariamente dallo stesso operatore. Essa può essere determinata risottoponendo all’analisi alcuni campioni già esaminati e determinando la deviazione standard dei risultati appaiati come è stato già descritto per la ripetibilità, oppure utilizzando opportune carte di controllo di qualità come sarà descritto nel Par. 1.3.4.
La ripetibilità e la riproducibilità della misura generalmente variano con il variare della concentrazione dell’analita nei campioni in esame. Pertanto nella pratica è preferibile esprimere la dispersione dei dati mediante il coefficiente di variazione (C.V. = 100 s / xm) ottenuto dal rapporto percentuale fra la deviazione standard e il valore medio. Il coefficiente di variazione è generalmente maggiore del 5% per quanto riguarda la riproducibilità, mentre valori relativamente più bassi vengono riportati per la ripetibilità della misura.
1.3.2. Accuratezza
L’accuratezza è definita dalla differenza fra la media dei valori ottenuti e il valore vero. Essa diviene così espressione di un errore sistematico nelle misura che porta a una sovrastima o a una sottostima dell’analita. Questo errore può essere imputabile ad uno sbaglio dell’operatore, ad una non raggiunta stabilizzazione della risposta dello strumento o ad una scarsa specificità del metodo.
A causa della difficoltà di determinare il valore vero, l’accuratezza di una analisi chimica talvolta sfugge alla possibilità di controllo da parte dell’analista. E’ tuttavia da tenere presente che, nella pratica clinica, la scarsa accuratezza dovuta ad una relativa non specificità del metodo non priva necessariamente del suo valore la misura quando questa è accompagnata dall’indicazione degli opportuni valori di riferimento, soprattutto se le sostanze interferenti non variano in modo marcato da individuo ad individuo.
1.3.3. Attendibilità
L’attendibilità di un metodo è la sua capacità di conservare la precisione ed la accuratezza nel tempo. Essa è particolarmente legata a quanto il metodo risente di modeste variazioni della tecnica, come quelle che si possono avere quando cambia l’operatore preposto alla misura o quando il metodo viene utilizzato in un nuovo laboratorio. La "robustezza" di un metodo (intesa come la capacità di non risentire di queste variazioni) può essere studiata introducendo deliberatamente dei piccoli cambiamenti nella procedura e confrontando i risultati.
1.3.4. Controllo dell’errore
Ogni laboratorio di analisi deve mettere in atto una serie di provvedimenti finalizzati a garantire la sicurezza della qualità dei risultati (Quality Assurance). Devono così essere scrupolosamente seguite tutte quelle norme necessarie per ridurre al minimo gli errori sia durante la fase preanalitica, analitica e postanalitica.
Fra gli errori che non attengono alla fase analitica, bisogna ricordare la possibilità di uno scambio di campione o di uno sbaglio nell’esecuzione del prelievo o nella conservazione del materiale biologico (di ciò verrà discusso nei paragrafi riguardanti le singole metodiche di analisi). Per quanto riguarda la fase analitica, la sicurezza della qualità dei risultati si basa su un controllo di qualità interno e su una valutazione esterna della qualità.
1.3.4a. Controllo di qualità interno
Il controllo di qualità interno è quella operazione di controllo della qualità analitica che ciascun laboratorio attua indipendentemente, generalmente mediante l’uso di uno o più standard. Il risultato del controllo deve essere disponibile in tempo reale, per permettere un giudizio su base statistica circa l’accettabilità dei risultati ottenuti nella serie analitica.
Lo standard può essere costituito (1) da una quantità pesata della sostanza in esame, chimicamente pura e disciolta in un volume appropriato (standard volumetrico primario), (2) da una soluzione contenente il componente oggetto dell’analisi, purificato fino ad un livello operativamente accettabile ma suscettibile di miglioramento qualora venissero ad essere disponibili nuovi metodi di separazione, la cui concentrazione è determinata non per pesata ma mediante metodi analitici di provata affidabilità (standard clinico secondario), o (3) da un campione di complessa composizione le cui caratteristiche fisico-chimiche simulano quelle del campione in esame (campione di riferimento).
Al posto dello standard, quando questo non è disponibile, si può utilizzare la media di tutte le analisi eseguite con un dato metodo (eventualmente, dopo aver eliminato i valori che cadono fuori certi limiti). Ciò si basa sul presupposto che le variazioni dalla media ottenute in serie successive di campioni relativamente omogenei devono essere trascurabili purché la media stessa interessi un numero sufficientemente elevato di determinazioni (almeno 100-200). La media può essere determinata sui campioni di un giorno o anche di più giorni successivi se il numero giornaliero è insufficiente.
Il controllo di qualità interno si attua in pratica inserendo
fra le analisi uno o più standard ed il bianco ogni volta
che si esamina una nuova serie di campioni sconosciuti. I dati
relativi a questi controlli vengono riportati in funzione del
tempo su un grafico cartesiano. Per facilitare la lettura del
grafico, si tracciano delle linee orizzontali in corrispondenza
del valore medio e dei valori che si discostano da esso di ±2 s e di ±3 s.
Queste ultime linee costituiscono, rispettivamente, il limiti di
guardia e di intervento per il controllo di qualità. Il metodo
è da considerarsi fuori controllo quando un valore dello standard
cade oltre il limite di intervento, più di 6 valori consecutivi
si trovano da uno stesso lato rispetto al valore medio o più di
6 valori consecutivi evidenziano una tendenza progressiva all’aumento
o alla diminuzione
![]() .
.
1.3.4b. Valutazione esterna della qualità
La valutazione esterna della qualità è un programma collaborativo avente lo scopo fondamentale di stabilire la comparabilità fra i risultati ottenuti nei vari laboratori. La gestione della valutazione della qualità è sotto il controllo di un ente esterno che invia ai singoli laboratori uno o più campioni di riferimento senza indicare la concentrazione degli analiti da determinare e che si fa carico dell’analisi statistica dei risultati. I dati dei singoli laboratori vengono confrontati con i "valori attesi" ottenuti (1) facendo analizzare il materiale da un numero sufficientemente elevato di laboratori di riferimento, che per competenza ed impiego di metodiche adatte sono da considerarsi i migliori, o (2) utilizzando i valori di consenso, cioè i valori medi ricavati da tutti i partecipanti al programma di valutazione esterna, tenuto conto delle differenti metodiche usate. L’ente coordinatore invia il risultato della propria analisi statistica agli interessati, mettendo in evidenza mediante opportuni istogrammi le differenze fra i dati del singolo laboratorio e gli altri e dando un giudizio sulla qualità dell’analisi.
La difficoltà maggiore che si incontra nell’organizzare la valutazione esterna della qualità è certamente di stabilire un rapporto di assoluta fiducia affinché il controllo possa essere utile ed efficace. Bisogna così evitare che i campioni della valutazione esterna siano trattati in modo speciale rispetto agli altri per timore di sanzioni nel caso di un giudizio negativo. Un altro rischio è che gli analisti che partecipano al programma di valutazione, invece di identificare la causa delle discrepanze nei risultati, siano portati ad alterare i sistemi di calibrazione nello sforzo di ottenere valori vicini a quelli attesi, ottenendo il risultato perverso di produrre valori inaccurati per i sieri dei pazienti.
1.4. Interferenze nelle analisi chimico cliniche
Una sostanza presente nel campione interferisce nell’analisi chimico clinica quando è capace di alterare il corretto valore dei risultati per un dato analita. Le sostanze interferenti possono essere, per quanto riguarda la propria origine, endogene o esogene e, in quest’ultimo caso, possono essere già contenute nel campione biologico all’atto del prelievo o venire aggiunte in un secondo momento. L’importanza di questo tipo di errori sulla qualità dei risultati è desumibile sia dalla loro frequenza sia dalla ricaduta negativa sull’utilizzo clinico del dato analitico. Per quanto riguarda i farmaci, si è potuto ad esempio osservare che le interferenze ad essi dovute sono più frequenti quando aumenta il numero di sostanze assunte dal paziente: la somministrazione contemporanea di numerosi farmaci ha un’alta probabilità di alterare in una qualche misura tutte le analisi. Di più difficile valutazione è l’impatto negativo sul paziente. Molte delle inconsistenze fra i dati analitici e il quadro clinico sono però certamente dovute a questo tipo di errori.
1.4.1. Sostanze interferenti endogene
Le interferenze dovute a sostanze endogene sono ascrivibili a condizioni di emolisi, iperlipemia, bilirubinemia, paraproteinemia o ad altri fattori.
L’emolisi del campione può essere dovuta ad una intrinseca fragilità delle emazie o ad un loro danneggiamento durante il prelievo o il trasporto (per insulto meccanico, congelamento, shock osmotico, aggiunta di detergenti o conservazione in un mezzo privo di glucosio). Il siero non appare emolizzato finché la concentrazione di emoglobina non è superiore a 0,2 g/L (vedi Par. 7.2); tuttavia i sieri itterici possono apparire non emolizzati anche quando sono presenti concentrazioni più elevate di emoglobina. L’emoglobina interferisce nelle analisi che utilizzano l’NAD(P)H come cromoforo in quanto essa inizia ad assorbire attorno i 340 nm (la metemoglobina, invece, non assorbe a questa lunghezza d’onda). L’emoglobina, inoltre, causa una sottostima della lipasi sierica e falsa in varia misura la determinazione della bilirubina (vedi Par. 5.2.1) e della teofillina (vedi Par. 13.3i). Altri composti presenti in alte concentrazioni nel globulo rosso (magnesio, potassio, ferro, lattico deidrogenasi, aspartato ed alanina aminotransferasi) possono dare luogo ad una sovrastima dell’analita nel siero nel caso di emolisi del campione. La sovrastima della creatina chinasi in campioni emolizzati è invece dovuta all’adenilato chinasi eritrocitaria che determina un aumento spurio di ATP nella miscela di reazione usata per la determinazione della chinasi (vedi Par. 9.4.1).
Grosse micelle lipidiche possono interferire nell’analisi in quanto aumentano la torbidità e la diffrazione del campione o causano una ripartizione dell’analita fra la fase acquosa e la fase apolare. Un aumento della torbidità incrementa il valore del bianco e riduce perciò l’intervallo operativo dei metodi colorimetrici; ulteriori errori possono essere causati da variazioni della torbidità stessa nel corso della misura per effetto dei reagenti aggiunti (ad esempio, nella determinazione enzimatica della trigliceridemia la torbidità diminuisce a seguito dell’aggiunta della lipasi, vedi Par. 3.5.1b). Una abnorme diffrazione del campione interferisce con i metodi nefelometrici. Il fatto che lo ione sodio non sia solubile nelle micelle lipidiche può essere causa di una condizione di pseudoiponatriemia quando questo analita viene determinato rispetto al volume totale del campione, mentre valori corretti sono ottenibili con misure che tengono conto della sola fase acquosa (ad esempio mediante l’uso di elettrodi selettivi, vedi Par. 12.1.1).
La bilirubina in elevate concentrazioni, per il forte assorbimento nell’intervallo tra 400 e 450 nm, può interferire in numerose reazioni colorimetriche. La bilirubina determina inoltre una sottostima della creatinina nella reazione di Jaffé (vedi Par. 6.4.1a) e, reagendo con intermedi delle reazioni che coinvolgono il perossido d’idrogeno, interferisce nei saggi enzimatici in cui è utilizzata la perossidasi per lo sviluppo del colore.
Le paraproteine possono essere presenti in circolo in caso di mieloma multiplo o di malattie simili. A volte la loro presenza determina un aumento considerevole della viscosità del siero, alterando la corretta misura volumetrica del campione. Le paraproteine possono interferire con il dosaggio del fosfato inorganico, quando è utilizzato il metodo all’ammonio molibdato (vedi Par. 12.6.1), o con il dosaggio dell’urea, quando è utilizzato il metodo all’o-ftalaldeide (vedi Par. 6.2.1a). Le immunoglobuline possono inoltre legare altre componenti plasmatiche. Il loro legame ad enzimi determina la comparsa di macroenzimi che sono generalmente catabolizzati più lentamente dall’organismo o che possono essere alterati nelle loro proprietà catalitiche (vedi Par. 9.3). Le immunoglobuline possono inoltre complessare la tiroxina causandone una sottostima quando è misurata con metodiche RIA (radio immuno assay) o una sovrastima in altri casi (vedi Par. 10.4.1a). A causa delle omologie strutturali, le immunoglobuline ad alte concentrazioni possono interferire nel dosaggio delle b-endorfine.
Molte altre sostanze endogene possono causare interferenze. Ad esempio la determinazione del cloruro mediante elettrodi selettivi risente della presenza di bicarbonato: si ha una sottostima dell’analita quando il bicarbonato è basso (< 15 mmol/L) e una sovrastima quando è alto (> 40 mmol/L) (vedi Par. 12.3.1). I corpi chetonici e le proteine interferiscono nella reazione di Jaffé per la determinazione della creatinina, ma in misura variabile a seconda della metodica usata (vedi Par. 6.4.1a).
1.4.2. Sostanze interferenti esogene
Le interferenze dovute a fattori esogeni non sono spesso facilmente prevedibili. I risultati delle analisi possono essere inficiati per la presenza di sostanze di derivazione farmacologica, additivi o per altre cause minori.
I farmaci, essendo stati progettati per essere attivi dal punto di vista biologico, hanno un’alta probabilità di reagire con gli analiti o con i reagenti usati in chimica clinica. Essi possono interferire come tali o dopo aver subito una serie di modificazioni metaboliche (generalmente a livello dei microsomi epatici) consistenti in reazioni di ossido-riduzione, idrolisi, esterificazione o coniugazione con acido glucuronico, glicina o solfato. Le interferenze causate dai prodotti metabolici dei farmaci sono più difficili da prevedere in quanto la loro natura e concentrazione non sono sempre note. Anche le modificazioni chimiche operate dall’industria sul farmaco per alterarne le proprietà biologiche possono modificare il grado di interferenza nei saggi biochimici. Ad esempio, la ciproeptadina (un antistaminico usato in alcune forme allergiche o come stimolante dell’appetito) interferisce nel dosaggio degli antidepressivi triciclici a cui è strutturalmente simile. La fenitoina (un antiepilettico usato anche come antiaritmico e, più raramente, nel trattamento della nevralgia trigeminale) e la 5-(p-idrossifenil)-5-fenilidantoina, suo principale metabolita, danno luogo a falsi positivi nei metodi intesi a svelare la presenza di barbiturici (vedi Par. 13.1a). La furosemide (un diuretico usato nella terapia degli edemi di varia origine e nell’ipertensione arteriosa) di per sé non interferisce con la reazione di Jaffé per la creatinina, ma un suo metabolita causa una sottostima di questo analita (vedi Par. 6.4.1a). La lidocaina (un anestetico locale e di superficie usato anche nel trattamento delle tachiaritmie ventricolari) interferisce con la determinazione enzimatica della creatina utilizzante la sarcosina ossidasi (vedi Par. 6.3.1a) in quanto viene metabolizzata dal fegato in 4-idrossi-2,6-xilidina e N-etilglicina, che è un composto chimico molto simile alla sarcosina.
Altre interferenze possono essere dovute a sostanze aggiunte al campione per renderlo incoagulabile (eparina, EDTA, citrato, ossalato), bloccare i processi glicolitici (fluoruro, iodoacetato), lubrificare la siringa (glicerolo) o sigillare il tappo della provetta (silicone). E’ stato visto che gli anticoagulanti e gli antiglicolitici, così come le sostanze inquinanti contenute nel silicone, possono interferire con numerosi metodi di analisi. Una contaminazione del campione con silicone può inoltre alterare la determinazione dello ione magnesio mediante elettrodi selettivi (vedi Par. 12.5.1). Il glicerolo può interferire nel dosaggio dei grassi neutri quando questo si basa proprio sulla determinazione quantitativa del glicerolo ottenuto per idrolisi del trigliceride (vedi Par. 3.5.1).
A volte il trattamento del campione può falsare i risultati delle analisi senza che sia possibile attribuire ciò alla presenza di una specifica sostanza chimica. Rientrano in questo gruppo di cause di errore i cambiamenti fisici indotti dalla liofilizzazione e dal congelamento. Altre interferenze possono essere attribuibili alla presenza di antifermentativi (azide sodica, antibiotici) e di stabilizzanti (ditiotreitolo, acetilcisteina). Un’altra possibilità è che sostanze interferenti siano contenute nei campioni usati per la calibrazione degli strumenti. Molti degli standard sono infatti ricostituiti a partire da plasma defibrinato arricchito (o depauperato) di alcuni suoi costituenti. Questi materiali contengono perciò spesso alte concentrazioni di metaboliti o farmaci che potrebbero essere causa di interferenze, falsando tutti i risultati ottenuti successivamente ad un particolare processo di calibrazione.
1.5. RACCOLTA DEI CAMPIONI DA ANALIZZARE
Il prelievo è una manovra diretta a raccogliere il materiale da analizzare e si prefigge tre obiettivi fondamentali: (1) ottenere un campione appropriato per volume, modalità di raccolta e caratteristiche analitiche; (2) limitare al massimo la variabilità delle condizioni di ottenimento del campione; (3) minimizzare i fastidi ed i rischi per il paziente.
Nel caso delle più comuni analisi eseguite su campioni di urina e di sangue venoso, di solito non è necessario ottenere un formale assenso orale o scritto da parte del paziente, che comunque deve sempre essere preventivamente informato sulla natura e sulla finalità dell’intervento medico. Il consenso del paziente deve essere ottenuto invece in forma esplicita nel caso di procedure più invasive quali, ad esempio, l’esame del midollo osseo, la toracocentesi, la puntura lombare e il prelievo arterioso. Ogni atto medico eseguito sul paziente ed ogni risultato di analisi devono essere ritenuti confidenziali e protetti da indiscrezioni. Tutto il personale del laboratorio deve assumere a stile di comportamento la riservatezza ed a norma di legge morale il segreto professionale.
Saranno descritte in dettaglio le più comuni tecniche di prelievo di interesse chimico-clinico, mentre per altre metodiche verranno dati solo dei brevi cenni, rimandando una trattazione più completa a testi specialistici.
1.5.1. Prelievi di sangue
Il prelievo ematico può avvenire secondo tre diverse modalità e riguardare il sangue periferico, il sangue venoso o il sangue arterioso. Il sangue prelevato può essere lasciato sierare o può essere reso incoagulabile mediante l’aggiunta di opportuni additivi.
1.5.1a. Sangue periferico
Il sangue periferico o capillare è una mescolanza di sangue arterioso, venoso e capillare e di liquidi intracellulari ed interstiziali, la cui composizione risente (1) del flusso di sangue verso la cute, (2) della composizione del sangue venoso a livello cutaneo e (3) della proporzione relativa fra sangue arterioso e venoso nel punto del prelievo.
Il prelievo di sangue periferico mediante puntura cutanea rappresenta una tecnica di elezione nei neonati e nella prima età pediatrica, sia perché impone una riduzione dei volumi di sangue da prelevare, sia perché meno invasivo e traumatico per il piccolo paziente. Questa tecnica è applicabile anche a pazienti adulti gravemente ustionati, a soggetti fortemente obesi o con tendenze trombotiche che devono ripetere frequentemente alcuni semplici test di laboratorio, come ad esempio la determinazione della glicemia (vedi Par. 2.1.2). Prelievi attraverso puntura cutanea venivano inoltre in genere usati in ricerche in campo ematologico su strisci di sangue; tuttavia, l’introduzione degli strumenti per la conta elettronica degli elementi corpuscolati su sangue venoso sta progressivamente rendendo obsoleta questa tecnica di prelievo (vedi Par. 7.1).
Le sedi elettive per il prelievo cutaneo sono (1) la superficie plantare laterale o mediale del tallone e (2) la superficie palmare (o plantare) di una falange distale. La puntura del tallone è raccomandata nei bambini fino al primo anno di età, prima che comincino a camminare, e va eseguita nella porzioni più mediali o più laterali della superficie plantare, comprendenti la porzione mediale ad una linea che partendo dalla metà dell’alluce raggiunge il tallone e la porzione laterale ad una linea che partendo dalla zona fra il quarto e il quinto dito raggiunge il tallone. La puntura del polpastrello di un dito, generalmente l’indice o il medio, è riservata ai bambini di oltre i 18 mesi ed agli adulti in quanto nei neonati (specie se prematuri) è facile provocare una lesione dell’osso (osteocondriti e, più raramente, gangrena) a causa della minore distanza di questo tessuto dalla superficie cutanea. La puntura del polpastrello deve essere eseguita al centro della falange distale (la distanza fra la superficie cutanea e l’osso nella zona superiore ed inferiore è la metà di quella esistente al centro) e l’incisione non deve superare i 3 mm di profondità.
Poiché si potrebbe incontrare una qualche difficoltà a raccogliere una quantità sufficiente di sangue, specialmente nel periodo freddo dell’anno, la parte da pungere deve essere preparata accuratamente riscaldandola per ravvivare la circolazione (nel caso del tallone, ciò può essere ottenuto avvolgendolo in compresse caldo-umide). La disinfezione deve essere praticata con garza o cotone preferibilmente imbevuti in alcool al 70%, che è la concentrazione ottimale per l’effetto germicida ed evapora facilmente senza provocare un raffreddamento della cute. Prima di procedere all’incisione è necessario attendere che la cute sia perfettamente asciutta per evitare che il sangue scoli di lato rendendo più difficile la raccolta. Per ottenere una maggiore quantità di sangue, è consigliabile spalmare la zona con una pomata al silicone che, evitando il contatto del sangue con la superficie cutanea, ritarda l’emostasi. L’incisione viene generalmente praticata con aghi ipodermici o lancette monouso, che possono essere anche montate su dispositivi con scatto a molla, dando un colpetto rapido e preciso e facendo in modo che l’incisione sia perpendicolare all’asse dei solchi digitali. E’ consigliabile scartare la prima goccia di sangue che contiene maggiori quantità di liquido interstiziale. Terminata la raccolta del sangue l’emostasi è generalmente rapidamente assicurata da una leggera pressione con una compressa di garza sterile.
Il sangue può essere raccolto in capillari di vario diametro oppure in microprovette di polietilene con capacità di circa 0,4 mL, contenenti anticoagulanti o un separatore per meglio ottenere il siero e fornite di un beccuccio per la raccolta del sangue dall’area incisa (completato il prelievo, il beccuccio viene rimosso e la provetta viene chiusa con un tappo di colore diverso a seconda del tipo di test da eseguire). Talora il campione può essere raccolto direttamente su speciali carte da filtro per eseguire particolari test di screening o fatto adsorbire su strisce reattive per una determinazione colorimetrica semiquantitativa di un analita.
1.5.1b. Sangue venoso
La relativa facilità di esecuzione e la scarsa invasività per il paziente rendono il prelievo venoso la fonte principale di campioni da analizzare nel laboratorio clinico. La composizione del sangue venoso non è costante ed omogenea in tutto l’organismo, come nel caso del sangue arterioso, perché essa può variare a seconda dell’attività metabolica dell’organo o del tessuto da cui il sangue è refluo. Tuttavia, nella maggioranza dei casi il prelievo venoso può ugualmente ritenersi rappresentativo della concentrazione di gran parte degli analiti.
Il prelievo venoso viene comunemente effettuato scegliendo una vena superficiale della faccia anteriore del braccio. Quando il prelievo brachiale si rivela impossibile, si può passare al prelievo venoso ai piedi o alle gambe, sede quest’ultima da evitarsi nel caso di pazienti con insufficienza cardiovascolare, diabete o malattie del circolo. Sono da evitarsi, in quanto non rappresentativi, i prelievi in aree ustionate o nelle quali sono presenti degli ematomi, i prelievi dal lato di una mastectomia, quando vi sia una stasi linfatica, ed i prelievi in zone dove sono presenti fistole o cannule.
Nella maggior parte dei casi è possibile individuare immediatamente la vena da utilizzare per il prelievo. E’ ovvio che si scelgono le vene più grosse e prominenti che al tatto appaiono "piene": solitamente la prima scelta cade sulla vena cubitale mediana, secondariamente si può ripiegare sulla cefalica che, rispetto alla basilica e ad altre, collassa e va incontro a rottura meno frequentemente (Fig. 1.4). Vene "difficili" possono occasionalmente essere riscontrate in soggetti normali, ma, più comunemente, sono tipiche dei pazienti obesi, cardiopatici e oncologici, che hanno ricevuto cicli di chemioterapia o sono stati sottoposti a lunghe e frequenti terapie endovenose. Se non si trova immediatamente una vena, si può invitare il paziente a chiudere e riaprire alcune volte la mano o ad abbassare il braccio per sfruttare la pressione idrostatica. In qualche caso si può provare a massaggiare l’avambraccio dal basso verso l’alto o a ricorrere al riscaldamento del lato volare dell’avambraccio. Nei casi particolarmente difficili, si può ricorrere alla applicazione di un laccio che, tuttavia, non può essere mantenuto in sede per oltre 2-3 minuti e deve essere comunque tolto almeno 2 minuti prima del prelievo, per non alterare il valore dei metaboliti plasmatici. La ricerca, non solo visiva, va affidata soprattutto alla sensibilità tattile della punta del polpastrello dell’indice al fine di riconoscere la vena che viene percepita come un tubicino elastico, di consistenza chiaramente differente dai tessuti circostanti, del quale bisogna valutare la profondità, la direzione e il calibro. In generale è sconsigliabile utilizzare vene troppo superficiali o troppo fragili che saranno in grado di fornire solo poche gocce di sangue e che, rompendosi con grande facilità, provocheranno vaste e demoralizzanti soffusioni emorragiche. In ogni caso è imperativo non tentare la puntura se non si è individuata con sicurezza la vena a costo di prolungare la ricerca.
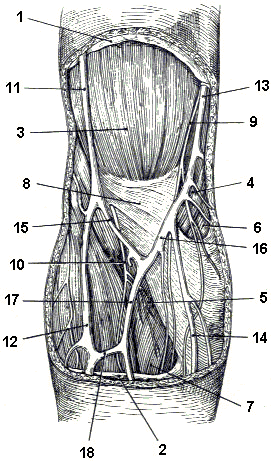
Fig. 1.4. Regione anteriore del gomito destro (piano superficiale). 1: fascia brachiale; 2: fascia antibrachiale; 3: muscolo bicipite brachiale; 4, 5: muscolo pronatore rotondo; 6, 7: muscolo flessore radiale del carpo; 8: lacertosus fibrosus; 9: nervo cutaneo mediale dell’avambraccio; 10: nervo cutaneo laterale dell’avambraccio; 11: vena cefalica del braccio; 12: vena cefalica dell’avambraccio; 13: vana basilica del braccio; 14: vena basilica dell’avambraccio; 15: vena mediana cefalica; 16: vena mediana basilica; 17: vena cubitale mediana; 18: ramo anastomotico.
Nei bambini le vene alla piega del gomito non sono visibili nella maggioranza dei casi, ma, se la tensione del laccio è corretta, sono chiaramente apprezzabili alla palpazione come cordoncini duro-elastici disposti longitudinalmente. Il loro diametro è ovviamente più piccolo che nell’adulto, ma generalmente si tratta di vasi che hanno pareti meno fragili e che sono ben fissi nei tessuti circostanti. Nei casi, piuttosto rari, in cui non si riesce ad apprezzare alcuna vena in corrispondenza della piega del gomito, si può ricorrere alla puntura della giugulare superficiale che appare in genere evidente lateralmente al collo se il piccolo paziente viene tenuto con il capo leggermente rovesciato controlateralmente e il cui turgore è accentuato dal pianto. Occorre tuttavia agire con particolare cautela, perché la parete della giugulare, al contrario delle vene dell’avambraccio, è piuttosto fragile. La puntura del seno sagittale attraverso la fontanella mediana è generalmente sconsigliabile per il rischio di ematoma e trombosi tardive di estrema gravità. Bisogna inoltre ricordare che in molti casi è possibile ricorrere a micrometodi di analisi per i quali è sufficiente una quantità limitata di sangue capillare, che può essere raccolta mediante una semplice puntura cutanea.
Una volta individuata la vena, si procede alla disinfezione della parte prescelta mediante un batuffolo di cotone o un tampone di garza intriso nel disinfettante (generalmente una soluzione idro-alcolica capace di evaporare piuttosto rapidamente in quanto è opportuno che la pelle sia asciutta al momento del prelievo). Viene quindi applicato un laccio, costituito di solito da un tubo di gomma morbida ed elastica da stringere attorno al braccio a circa 10 cm al di sopra del punto di prelievo, introducendo un’ansa in prossimità di uno dei due capi, in modo che il laccio stesso possa venire sciolto rapidamente se necessario, e verificando che il polso arterioso a valle continui ad essere percepibile. Si procede infine al prelievo di sangue mediante un ago montato direttamente sulla siringa, un ago a farfalla (butterfly) connesso alla siringa mediante un tubicino o un sistema per prelievi sotto vuoto (vacutainer).
Il diametro dell’ago a perdere viene indicato con numeri convenzionali, detti gauge, che sono in genere compresi fra 18 e 25 e il cui valore è inversamente proporzionale alla sezione dell’ago. Il calibro dell’ago deve essere adeguato a quello della vena dalla quale si intende prelevare il sangue. Quando si prevede che la vena sia abbastanza "facile", è bene usare aghi di sezione piuttosto grande perché il dolore provocato dalla puntura non dipende dal diametro dell’ago, se è bene affilato, mentre il sangue scorre più facilmente quando la vena è stata bene incannulata. Se la vena è visibile per un tratto breve o se è stata individuata solo per palpazione, è consigliabile pungere al di sotto della zona visibile o palpabile per evitare di andare oltre la vena. L’ugnatura dell’ago deve essere rivolta verso l’alto per evitare che il lume venga ostruito mentre l’ago penetra. L’asse dell’ago deve corrispondere al percorso della vena ed avere una inclinazione di circa 15° in modo da penetrare sicuramente attraverso la parete superficiale della vena. La mano che guida l’ago avverte subito sotto la cute una sensazione di resistenza alla penetrazione cui segue una sensazione di vuoto, quando l’ago è entrato in vena. A questo punto è bene spingere ancora un poco in avanti l’ago, dopo averne ridotto leggermente l’inclinazione, per fare in modo che la vena venga bene incannulata. Quando la mancanza di vene palpabili e bene identificabili costringe l’operatore ad usare vasi superficiali o molto sottili, è necessario scegliere aghi di calibro minore per evitare di ledere il vaso provocando la contrazione delle sue pareti e la formazione di soffusioni emorragiche. In questi casi bisogna ridurre l’inclinazione dell’ago rispetto la superficie dell’arto e penetrare nella cute con delicatezza il più lontano possibile dalla vena cercando di seguirne il percorso con l’ago.
Gli aghi del vacutainer sono formati da due segmenti collegati da un cono di plastica con filettatura: il segmento più lungo costituisce l’ago vero e proprio, il più corto serve a forare il tappo di gomma che mantiene il vuoto nella provetta, mettendo in comunicazione il lume della vena con la provetta stessa (Fig. 1.5). E’ possibile inserire sullo stesso ago differenti provette per la raccolta del sangue in varie condizioni in quanto una guaina di gomma arresta il flusso di sangue durante il cambio di provetta. Le provette, di plastica o di vetro, hanno tappi di differenti colori in modo da informare l’operatore sulla "quantità di vuoto precalibrato" e sulla eventuale presenza di anticoagulanti. Il vacutainer offre molti vantaggi soprattutto prevenendo l’emolisi perché con questo sistema si esercita una aspirazione costante e continua, si evita che l’aria passi tra il cono dell’ago e il cono della siringa e si elimina la fase si svuotamento della siringa in provetta. Con la siringa, tuttavia, si può essere sempre sicuri che la vena è stata correttamente incannulata quando compare un fiotto di sangue nel cono della siringa stessa, mentre con il vacutainer questa sicurezza si ha solo dopo aver messo in comunicazione la provetta con l’ago. Rispetto al vacutainer e all’ago montato direttamente sulla siringa, la butterfly ha il vantaggio di poter essere facilmente mantenuta nella posizione dovuta una volta introdotto l’ago in vena, ma il piccolo calibro e il lungo e sottile tubicino di raccordo impongono di esercitare sul pistone della siringa una aspirazione più energica che può provocare l’emolisi del sangue.
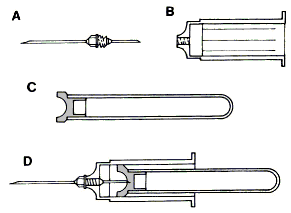
Fig. 1.5. Vacutainer. A: ago; B: portaprovette; C: provetta sotto vuoto; D: strumentario montato per prelievi sotto vuoto
Il laccio va sciolto durante l’effettuazione del prelievo o appena questo è terminato e comunque prima che venga estratto l’ago. L’ago viene estratto con decisione e delicatezza e l’emostasi viene facilitata comprimendo sulla ferita un batuffolo di cotone ed invitando il paziente a flettere l’avambraccio ed a sollevare il gomito al di sopra del cingolo scapolare, per sfruttare il noto effetto vasocostrittore conseguente allo stiramento del plesso brachiale. Avvenuta l’emostasi, la ferita viene detersa con un disinfettante, mentre è inutile l’applicazione di un cerotto medicato. Nei rari casi in cui il sanguinamento non è cessato dopo un quarto d’ora, è necessario intervenire con la terapia indicata nei casi di diatesi emorragica. La comparsa di un ematoma nel punto del prelievo può essere dovuta ad una qualche manovra errata nella ricerca della vena, alla mancata rimozione del laccio prima dell’estrazione dell’ago o ad una insufficiente compressione della ferita quando l’ago è stato estratto. Una complicazione relativamente frequente è una crisi lipotimica, che recede spontaneamente ponendo il paziente in posizione supina. Una rara complicazione tardiva è rappresentata dalla trombosi del vaso, che si verifica soprattutto quando la vena è stata incannulata con molta frequenza. L’insorgenza di una tromboflebite o il contagio con il virus dell’epatite sono evenienze improbabili da quando sono state universalmente adottate siringhe ed aghi monouso.
Se il prelievo è stato eseguito con una siringa, l’operatore deve immediatamente procedere al trasferimento del sangue nelle varie provette. Disinnescato l’ago, si appoggia il cono della siringa sulla parete della provetta e, spingendo lentamente il pistone, si espelle la quantità opportuna di sangue lungo la parete della provetta, ripetendo con calma l’operazione con tutte le provette necessarie. Le provette contenenti additivi vengono chiuse e rovesciate gentilmente 2-3 volte per assicurare la distribuzione omogenea di composti senza formare schiuma.
1.5.1c. Sangue arterioso
Un campione di sangue arterioso è essenziale per valutare la tensione dei gas ed in particolare quella dell’ossigeno e dell’anidride carbonica, come indice della funzionalità respiratoria, e del monossido di carbonio, nei casi di sospetta intossicazione. Inoltre, il campione arterioso diventa preferibile a quello venoso quando vi siano alterazioni della circolazione sistemica o locale. Tuttavia, il prelievo arterioso non è solo tecnicamente più difficile da eseguire, ma soprattutto è più doloroso e pericoloso per il paziente.
I criteri di scelta della sede di prelievo si basano, più che per gli altri tipi di prelievo, su una buona conoscenza dell’anatomia e su un certo grado di addestramento che ponga in grado il medico di saper valutare il calibro e l’accessibilità dell’arteria, lo stato del tessuto periarterioso e la presenza di un circolo collaterale.
1.5.1d. Anticoagulanti
La maggior parte degli esami di laboratorio sono eseguiti su siero (vedi Par. 7) e l’uso degli anticoagulanti è attualmente limitato a pochi procedimenti analitici aventi particolari esigenze tecniche, come la determinazione dell’ammoniemia (vedi Par. 6.1.2), della glicemia (vedi Par. 2.1.2), dei fattori della coagulazione (vedi Par. 7.3.5), della conta degli elementi figurati del sangue (vedi Par. 7.1.6) o della velocità di eritrosedimentazione (vedi Par. 8.7).
Gli anticoagulanti sono sostanze che impediscono la coagulazione bloccando l’azione del calcio (ossalato, citrato, EDTA) o inibendo l’attività della trombina o di altri fattori della coagulazione (fluoruro, eparina).
Gli ossalati si legano al calcio plasmatico formando ossalato di calcio insolubile; il più usato è l’ossalato di sodio in soluzione 0,1 M, che viene diluito nel rapporto di 1:9 con il sangue. Il citrato di sodio, che forma con il calcio un complesso solubile, è generalmente disponibile in soluzione 0,11 M ed è diluito nel rapporto di 1:8. L’EDTA (etilendiamino tetracetato di sodio) forma con il calcio un chelato non ionizzato e viene usato alla concentrazione di 1-2 mg per mL di sangue sia in polvere sia come soluzione al 10%.
Il fluoruro di sodio (2 mg/mL) ostacola la coagulazione, in quanto lega il calcio ed interferisce nella cascata enzimatica dell’emocoagulazione, inibendo nel contempo la glicolisi. Per quest’ultimo motivo, il fluoruro di sodio è aggiunto assieme all’ossalato di potassio ai campioni che devono essere utilizzati per determinare la glicemia.
L’eparina costituisce un gruppo eterogeneo di glicosaminoglicani solforati con una struttura polimerica formata da unità disaccaridiche unite da legami a(1-4) glicosidici e un peso molecolare compreso tra 6000 e 40000 (il peso molecolare medio di gran parte delle preparazioni commerciali è di 12000 - 15000). Le singole unità disaccaridiche sono composte da D-glucosamina ed un acido uronico (acido D-glucuronico o acido L-iduronico; vedi Fig. 1.6) e presentano i gruppi aminici e parte dei gruppi ossidrilici legati all’acido solforico. L’eparina si lega all’antitrombina attraverso una sua sequenza pentasaccaridica costituita da due residui di acido uronico e tre D-glucosamine, accelerando l’inattivazione del fattore Xa e della trombina e prevenendo così la conversione del fibrinogeno in fibrina (vedi Par. 7.3.1h). L’eparina è un ottimo anticoagulante, anche se relativamente più costoso, che viene usato alla concentrazione di 0,2 mg (20 UI) per mL e, in genere, non interferisce in laboratorio con le determinazioni chimiche ed enzimatiche.
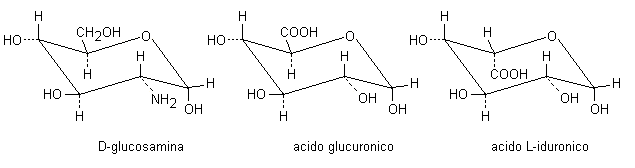
Fig. 1. 6. Struttura dei monosaccaridi costituenti l’eparina.
1.5.2. Prelievo di campioni d’urina
Gli esami che vengono compiuti sull’urina sono molteplici e le modalità del prelievo variano a seconda dei casi (vedi Par. 11). I campioni vengono generalmente classificati come estemporanei o temporizzati.
1.5.2a. Campioni estemporanei
Un campione estemporaneo di urine non si presta alla determinazione quantitativa dei vari costituenti, ma solo ad una loro valutazione semiquantitativa perché la concentrazione delle sostanze è fortemente influenzata dalla quantità di urina che viene emessa nell’arco della giornata. La valutazione del peso specifico consente tuttavia di correggere almeno parzialmente i risultati anomali dovuti ad una riduzione o ad un aumento della diuresi. Il campione ottenuto di prima mattina da un soggetto ancora a digiuno è generalmente di scelta in quanto rappresentativo della composizione dell’urina nell’arco della nottata.
Il campione da utilizzarsi per le analisi biochimiche viene raccolto in un recipiente non necessariamente sterile, ma pulito, ed inviato prontamente in laboratorio. Nel caso che sia necessario ottenere un campione rappresentativo della flora batterica delle vie urinarie, si ricorre alla tecnica del mitto intermedio. La tecnica consiste nell’evitare accuratamente la contaminazione del campione da parte di batteri delle mani, dei vestiti, della pelle e dei peli del perineo e delle secrezioni vaginali o prepuziali. Dopo una accurata pulizia con garze imbevute di antisettico, il paziente dirige il primo getto in un orinale e raccoglie la successiva urina in un contenitore sterile che deve essere chiuso immediatamente con un tappo che assicuri la tenuta completa.
1.5.2b. Campioni temporizzati
Le raccolte temporizzate più comuni sono rappresentate dalle urine delle 12 o delle 24 ore. Queste raccolte sono raccomandate per la determinazione di analiti le cui concentrazioni possono variare significativamente nel corso della giornata o a seconda dello stato di digiuno. Raccolte temporizzate più brevi sono utilizzate in ambito pediatrico.
La raccolta delle urine delle 24 ore si effettua invitando il paziente a svuotare la vescica ad un’ora determinata della giornata (preferibilmente al risveglio al mattino) ed a scartare questa urina. Successivamente, tutte le urine emesse di volta in volta sono raccolte in un apposito contenitore che viene conservato in frigorifero; la raccolta è conclusa aggiungendo nel contenitore l’urina emessa il giorno successivo all’ora corrispondente a quella dell’inizio della prova. Nel caso sia necessario aggiungere nel recipiente conservanti pericolosi, il paziente deve essere istruito accuratamente sulle precauzioni da adottare.
1.5.3. Prelievo di succo gastro-enterico e di feci
Il succo gastrico viene raccolto dopo intubazione nasale (tubo di Levin) od orale (tubo di Rehfus). Il paziente deve essere informato dell’esame che verrà eseguito e deve assicurare una buona cooperazione. Per la raccolta del succo duodenale, vengono usati vari tipi di tubi a doppia o tripla via (Diamond, Dreiling) il cui posizionamento deve essere guidato radioscopicamente; le secrezioni vengono aspirate mediante una pompa. I campioni sono raccolti in un adatto contenitore ed inviati immediatamente in laboratorio.
Le feci possono essere emesse in contenitori sterili reperibili in commercio o trasferite in essi dalla padella mediante una spatola. Nel lattante, le feci possono essere raccolte dal pannolino o direttamente dal retto mediante l’introduzione di un tampone. Poiché è estremamente difficile una raccolta esatta delle feci emesse nelle 24 ore, è preferibile prolungare la raccolta per 3 giorni, ricorrendo anche all’espediente di somministrare un colorante non assorbibile (carminio) all’inizio del periodo prestabilito e una dose di carbone alla fine, in modo da raccogliere le feci dal momento in cui inizia a comparire il colorante al momento che precede la comparsa del carbone.
1.5.4. Prelievo di liquido cefalo-rachidiano
Il liquor (o liquido cefalo-rachidiano) viene prelevato solitamente attraverso rachicentesi tra la terza e la quarta vertebra lombare o, meno comunemente, attraverso puntura cervicale, ventricolare o della cisterna. Il liquor deve essere raccolto in tre diverse provette sterili a chiusura completa (1) per gli esami chimico-clinici ed immunologici, (2) per gli esami microbiologici e (3) per il conteggio differenziale degli elementi figurati. Il campione va inviato immediatamente al laboratorio con procedure separate e prioritarie rispetto ad altro materiale biologico.
Normalmente il liquor è limpido, omogeneo, incolore, del tutto simile all’acqua ("acqua di roccia"). Lasciato a sé e centrifugato, non coagula e non lascia apprezzare alcun sedimento. L’ispezione del liquor permette di evidenziare la presenza di torbidità o alterazioni del colore che sono giudicate in base all’intensità con un numero progressivo di segni + da 1 a 4.
La torbidità del liquor, generalmente dovuta alla presenza di cellule in sospensione (> 400-500 per mL), può essere lieve, come nel caso di una meningite tubercolare, o franca, come nel caso di una meningite da piogeni. In condizioni patologiche, possono essere presenti dei coaguli di fibrina. Nel caso di meningite tubercolare o in altre forme di meningite, si può osservare la comparsa di un delicato reticolo di fibrina (reticolo di Mya), quando il liquor è lasciato perfettamente immobile per un periodo di 12-24 ore. In caso di gravi compressioni midollari con blocco degli spazi subaracnoidei, il liquor può andare incontro ad una rapida e massiva coagulazione per la presenza di elevate quantità di fibrinogeno e altre proteine e di elementi cellulari (sindrome di Froin).
Un colorito ematico può dipendere dalla puntura accidentale di un vaso durante il prelievo o da un versamento di sangue negli spazi subaracnoidei, avvenuto per cause patologiche: nel primo caso il sangue tende a diminuire man mano che il liquor fuoriesce, mentre nel secondo caso l’aspetto del liquor si mantiene costante durante tutta la raccolta. Un colorito giallo (xantocromico) può essere dovuto a malattie itterigene (ad es. la malattia emolitica del neonato, vedi Par.5.2) o ad una pregressa emorragia intracerebrale o subaracnoidea. Il liquor appare xantocromico circa 12 ore dopo l’episodio emorragico, si fa intensamente colorato dopo 2-4 giorni e ritorna incolore dopo 15-20 giorni.
La concentrazione delle proteine totali (vedi Par. 8.7.1b) nel liquor prelevato per puntura lombare è di 20-50 mg/dL nell’adulto e di 30-200 mg/dL nei neonati, mentre valori ancora più elevati sono riscontrabili negli immaturi. Nell’adulto, la concentrazione delle proteine totali è di 5-10 mg/dL nei ventricoli laterali e di 15-25 mg/dL nella cisterna magna. Aumenti della concentrazione proteica totale si possono trovare in numerose condizioni patologiche caratterizzate da un’alterazione della permeabilità della barriera ematoencefalica. Si indica con il termine di "dissociazione albumino-citologica" una particolare situazione nella quale sono aumentate nel liquor le proteine, mentre è normale il numero degli elementi figurati; questa situazione è caratteristica di una forma di polinevrite acuta (sindrome di Guillain-Barré), ma può riscontrarsi anche in altre polinevriti, tumori cerebrali, trombosi o blocco subaracnoideo.
Il quadro elettroforetico (vedi Par.
8.7.3) delle proteine liquorali
differisce da quello delle proteine sieriche per la presenza di una netta banda
prealbuminica (3-5% del totale), una maggiore quantità di β-globuline
(15-26% del totale) e una scarsa quantità di γ-globuline
(6-10% del totale); la frazione β è spesso sdoppiata
in due bande, per la presenza di una variante della transferrina con minore
mobilità elettroforetica (banda β2 o τ). Per interpretare il quadro elettroforetico del
liquor è sempre necessario esaminare anche il quadro elettroforetico del siero
dello stesso paziente. Esso può essere modificato per il passaggio di proteine
del plasma nel liquor a causa di un aumento della permeabilità della barriera
ematoencefalica o di una ostruzione meccanica degli spazi aracnoidei midollari
e/o essere influenzato dalla composizione delle proteine plasmatiche per un
aumento delle frazioni α1 e α2 (in caso di reazioni di fase acuta;
vedi Par. 8.2) o
della frazione γ (in caso di processi infiammatori
cronici o di cirrosi epatica; vedi Par. 8.7.3) o per la presenza di componenti monoclonali (vedi
Par. 8.6.2b). Nelle
sindromi infiammatorie interessanti il sistema nervoso centrale e, con
particolare frequenza, nei pazienti affetti da sclerosi a placche
![]() , si osserva un aumento delle γ-globuline con la presenza di bande oligoclonali
dovute alla sintesi di IgG a livello del tessuto nervoso, con una netta
dissociazione del quadro elettroforetico liquorale dal quadro elettroforetico
plasmatico
, si osserva un aumento delle γ-globuline con la presenza di bande oligoclonali
dovute alla sintesi di IgG a livello del tessuto nervoso, con una netta
dissociazione del quadro elettroforetico liquorale dal quadro elettroforetico
plasmatico
![]() .
.
La concentrazione di glucosio nel liquor (glicorrachia) è normalmete di poco inferiore a quella nel sangue (50-80 mg/dL; vedi Par. 2.1.2). Le variazioni della glicemia sono seguite con un ritardo di 1-3 ore da analoghe variazioni della glicorrachia. Diminuzioni della glicorrachia si osservano in caso di meningite (in modo più spiccato nelle meningiti purulente), tumori meningeali e infiltrazioni leucemiche.
La concentrazione dei cloruri nel liquor (clorurorrachia) è leggermente superiore a quella del sangue (115-130 mEq/L; vedi Par. 12.3.2). Una diminuzione della clorurorrachia (< 90 mEq/L) si verifica in modo particolare nella meningite tubercolare.
1.5.5. Prelievo di trasudati ed essudati in cavità fisiologiche
Campioni di liquido pleurico, pericardico e peritoneale sono raccolti mediante paracentesi in condizioni di sterilità e conservati in idonei contenitori che devono essere mantenuti 4°C ed inviati in laboratorio entro un’ora dal prelievo. L’esame microscopico comprende il conteggio differenziale dei leucociti, la conta degli eritrociti e l’esame citologico. L’esame chimico-clinico va eseguito sul sopranatante liberato dal materiale corpuscolato e dal coagulo. E’ raccomandabile eseguire in parallelo i medesimi esami su un campione di sangue prelevato simultaneamente al paziente.
Il liquido sinoviale è aspirato per artrocentesi su pazienti a digiuno da almeno 6-8 ore per ottenere un valido equilibrio con la concentrazione del glucosio nel sangue. La procedura raccomandata prevede una prima raccolta di 5-10 mL di liquido per l’esame microbiologico in provette contenenti eparinato di sodio, una seconda raccolta di 2-5 ml per l’esame microscopico in una provetta contenente lo stesso anticoagulante ed infine una terza raccolta, senza anticoagulante, per i test chimico-clinici.
1.6. APPENDICE: GRANDEZZE ED UNITA’ DI MISURA
Al fine di giungere ad una immediata comprensione dell’espressione
dei risultati, si è proceduto alla codificazione di un sistema
di unità di misura delle varie grandezze, unificato nella
definizione, nella nomenclatura e nella simbologia. Il sistema
attualmente adottato prende il nome di "Sistema
Internazionale di Unità di Misura"
![]() . Il Sistema Internazionale di
Unità di Misura si fonda su 7 grandezze e relative unità di
base (a cui devono essere aggiunte 2 grandezze e relative unità
supplementari, utili nel campo della geometria), individuate da
nomi, simboli e dimensione e ritenute sufficienti a misurare
tutte le grandezze che interessano la Fisica, la Chimica e la
Geometria (Tab. 1.IV)
. Il Sistema Internazionale di
Unità di Misura si fonda su 7 grandezze e relative unità di
base (a cui devono essere aggiunte 2 grandezze e relative unità
supplementari, utili nel campo della geometria), individuate da
nomi, simboli e dimensione e ritenute sufficienti a misurare
tutte le grandezze che interessano la Fisica, la Chimica e la
Geometria (Tab. 1.IV)
![]() .
.
Tab. 1.IV. Sistema Internazionale di Unità di Misura
| Grandezza | Unità | |||
| Nome | Simbolo | Dimensione | Nome | Simbolo |
| Grandezze ed unità di base | ||||
| Lunghezza | l | L | metro | m |
| Massa | m | M | kilogrammo | kg |
| Tempo | t | T | secondo | s |
| Corrente elettrica | I | I | ampere | A |
| Temperatura | T | Θ | kelvin | K |
| Quantitàdi sostanza | n | N | mole | mol |
| Intensità luminosa | Iv | J | candela | cd |
| Grandezze ed unità supplementari | ||||
| Angolo piano | α, β, γ, ... | uno | radiante | rad |
| Angolo solido | ω, Ω | uno | steradiante | sr |
Allo scopo di esprimere il risultato con l’appropriato
numero di cifre significative e in modo semplice quando il valore
numerico è troppo grande o troppo piccolo, è conveniente
utilizzare adatti multipli o sottomultipli moltiplicando l’unità
per potenze di 10 e indicando questa trasformazione attraverso
dei prefissi anteposti al nome e al simbolo dell’unità
stessa. E’ raccomandato l’uso dei fattori mostrati
nella Tab.1.V, che fanno ogni volta variare l’unità
di 3 ordini di grandezza, mentre è generalmente sconsigliato, ma
a volte tollerato, l’uso dei fattori 10 e 100. Così, un’escrezione
giornaliera urinaria di aldosterone può essere indicata
indifferentemente uguale a 55 · 10-9 mol (leggi: moli),
0,055 μmol (leggi: micromoli) o 55
nmol (leggi: nanomoli). Non è invece ammesso l’uso di più
prefissi contemporaneamente (ad esempio, non è corretto
scrivere 55 mμmol)
![]() .
.
Tab. 1.V. - Fattori decimali per indicare multipli e sottomultipli dell’unità
| Fattore | Prefisso | Simbolo | Fattore | Prefisso | Simbolo |
Fattori raccomandati |
|||||
| 1018 | exa | E | 10-18 | atto | a |
| 1015 | peta | P | 10-15 | femto | f |
| 1012 | tera | T | 10-12 | pico | p |
| 109 | giga | G | 10-9 | nano | n |
| 106 | mega | M | 10-6 | micro | μ |
| 103 | kilo | k | 10-3 | milli | m |
Fattori sconsigliati |
|||||
| 102 | etto | h | 10-2 | centi | c |
| 101 | deca | da | 10-1 | deci | d |
Le grandezze derivate si ottengono dal prodotto e/o dal quoziente delle grandezze di base e supplementari. La dimensione delle grandezze derivate è pari al prodotto e/o al quoziente delle dimensioni delle grandezze da cui derivano (Tab. 1.VI). Le unità delle grandezze derivate possono essere coerenti, quando sono prodotto e/o quoziente di sole unità di base e supplementari (cioè non moltiplicate per un fattore), o non-coerenti, quando sono prodotto e/o quoziente di unità di base e supplementari moltiplicate, tutte o in parte, per dei fattori numerici. L’uso di unità non coerenti è sconsigliato, ma per usi pratici alcune di queste unità sono permesse. Per comodità, molto spesso le unità delle grandezze derivate hanno un nome e un simbolo speciale.
Tab. 1.VI. Grandezze ed unità di maggiore interesse per la chimica clinica
| Nome (abbreviazione) | Simbolo | Dimensione | Unità |
|
| coerenti | non coerenti | |||
| Area (ar.) | A ; As ; S | L2 | m2 | |
| Attività catalitica (act.) | z | T -1N | mol/s ; kat | μmol/min ; U ; IU |
| Conc. catalitica (cct.) | b | L-3T -1N | kat/L ; U/L | |
| Conc. di massa (cms.) | ρ | L-3M | kg/L | |
| Conc. di numero (cnm.) | C | L-3 | 1/L | |
| Conc. di sostanza (cst.) | c ; [sostanza] | L-3N | mol/L ; M | |
| Densità-massa (dnms.) | ρ | L-3M | g/L | |
| Densità relativa (dnrel.) | d | uno | ||
| Molalità (molal.) | m | M-1N | mol/kg | |
| Numero di entità (nm.) | N | uno | ||
| Peso molecolare (pm.) | Mr | uno | ||
| Pressione (ps.) | p ; P | L-1MT -2 | Pa | mmHg |
| Temp. Celsius (tp.°C) | θ ; t | Θ | °C | |
| Tempo (tm.) | t | T | s | min ; h ; d |
| Volume (vl.) | V ; v | L3 | L ; l | |
| Volume specifico (vlspf.) | v | L3M-1 | L/g | |
Per quanto riguarda le grandezze di più frequente uso in
biochimica clinica, l’area e la molalità sono generalmente
espresse in unità coerenti. La temperatura viene usualmente
espressa, in modo difforme al Sistema Internazionale, come "temperatura
Celsius" (definita dall’equazione q/°C
= (T/K) - 273,15), tuttavia si deve tener presente che l’unità
di intervallo di "temperatura Celsius" è coerente in
quanto uguale al "kelvin". La pressione viene indicata
nell’unità coerente "pascal" (Pa = m-1·kg·s-2)
per la misura della pressione parziale dei gas nel sangue, mentre
l’unità tradizionale "millimetri di mercurio" (mmHg
= 133,322 Pa) viene usata soltanto per misurare la pressione del
sangue e di altri fluidi biologici. Per la misura dell’attività
catalitica, l’unità derivata coerente "mole al secondo"
(nome proposto: katal; simbolo: kat) dovrebbe sostituire l’unità
non-coerente "micromole al minuto" (nome proposto: Unità
Interanzionale; simbolo: IU o, più semplicemente, U)
corrispondente a 16,67 nkat, tuttavia l’uso dell’unità
non-coerente è ancora largamente diffuso. Il volume è
usualmente espresso nell’unità non-coerente "litro"
(L = 10-3 m3), così
come sono espresse in modo non-coerente alcune misure (come la
concentrazione, la densità-massa e il volume specifico) che
fanno riferimento a un volume unitario. Nella misura del tempo è
frequente l’uso delle unità non-coerenti "minuto"
(min = 60 s), "ora" (h = 3600 s) e "giorno" (d
= 86400 s). La densità relativa, il numero di entità e il peso
molecolare sono grandezze con dimensione unitaria e quindi
espresse mediante numeri puri. Bisogna ricordare infine che la
quantità o la concentrazione degli analiti dovrebbero essere
espresse rispettivamente in "quantità di sostanza" (unità:
mole) e in "concentrazione di sostanza" (unità: mole
al litro) quando è nota l’entità elementare dell’analita
stesso (molecola, ione, etc.) al fine di avere una immediata
percezione del rapporto stechiometrico in cui si trovano i
diversi componenti in un dato sistema, cosa che non si ottiene
con l’uso delle grandezze "massa" (unità:
kilogrammo) e "concentrazione di massa" (unità:
kilogrammo al litro)
![]() .
Le grandezze e unità di "massa" devono però essere
obbligatoriamente utilizzate nel caso di analiti a composizione
molecolare non ben definita o nel caso del dosaggio globale di
miscele di componenti, a meno che queste non vengano espresse
attraverso uno dei componenti molecolarmente ben definito.
.
Le grandezze e unità di "massa" devono però essere
obbligatoriamente utilizzate nel caso di analiti a composizione
molecolare non ben definita o nel caso del dosaggio globale di
miscele di componenti, a meno che queste non vengano espresse
attraverso uno dei componenti molecolarmente ben definito.
| aggiornamento: 26/07/11 |