|
|
Costantino Salerno |
|
|
Appunti di Biochimica Clinica |
|
|
2. METABOLISMO GLUCIDICO
Nei soggetti che seguono la comune dieta dei paesi
occidentali, il 40-60% del fabbisogno energetico totale dell’organismo
è soddisfatto dall’ossidazione del glucosio. Questo
rappresenta un substrato energetico pressoché esclusivo per il
sistema nervoso centrale in un soggetto ben alimentato. Altri
tessuti possono utilizzare sia il glucosio sia gli acidi grassi
ed i corpi chetonici (prodotti dalla parziale ossidazione degli
acidi grassi), ricorrendo in modo intercambiabile all’uno o
all’altro di questi substrati a seconda della loro
disponibilità nel sangue
![]() .
In generale, il glucosio è il principale substrato energetico
durante una attività fisica intensa e nelle prime 4-6 ore dopo
un pasto.
.
In generale, il glucosio è il principale substrato energetico
durante una attività fisica intensa e nelle prime 4-6 ore dopo
un pasto.
Le fonti di glucosio sono rappresentate dall’apporto
alimentare (durante la fase postprandiale) e dalla gluconeogenesi
e glicogenolisi (durante la fase postassorbimento). Il glucosio
viene assunto nell’organismo direttamente attraverso il
tratto digerente, dove si forma per idrolisi da diversi
carboidrati. Altri zuccheri eventualmente assorbiti nell’intestino
sono convertiti in glucosio dal fegato. Il glucosio viene inoltre
sintetizzato a partire dagli amino acidi glucogenici
![]() e dal glicerolo, lattato e
piruvato nel fegato e, in minore misura, in altri organi in cui
è attiva la gluconeogenesi, come il rene. La produzione epatica
giornaliera di glucosio è di 200-300 g, a seconda della sua
disponibilità negli alimenti e delle esigenze dell’organismo.
e dal glicerolo, lattato e
piruvato nel fegato e, in minore misura, in altri organi in cui
è attiva la gluconeogenesi, come il rene. La produzione epatica
giornaliera di glucosio è di 200-300 g, a seconda della sua
disponibilità negli alimenti e delle esigenze dell’organismo.
Il glucosio è immagazzinato sotto forma di glicogeno. Il glicogeno è depositato per la maggior parte nel fegato (60-130 g) e nel muscolo scheletrico (400-600 g), mentre è presente in minor misura in altri tessuti. Il glicogeno muscolare costituisce una riserva di substrato per uso locale, ma non può essere utilizzato per liberare glucosio in circolo. Questo viene invece rilasciato dal fegato ed assunto dalle cellule di quasi tutti gli altri organi e tessuti.
Nei tessuti, il glucosio è rapidamente convertito in glucosio 6-fosfato e metabolizzato attraverso quattro diverse vie: (1) convertito in piruvato e lattato attraverso la glicolisi anaerobia, (2) ossidato ad anidride carbonica ed acqua attraverso il ciclo degli acidi tricarbossilici, (3) ossidato a ribosio ed anidride carbonica attraverso la via degli esosi monofosfati, (4) convertito in acido glucuronico attraverso la via degli acidi uronici. Di consueto, il 10-20% del glucosio rilasciato dal fegato non viene ossidato completamente bensì convertito a lattato, piruvato, glicerolo ed aminoacidi, che tornano poi al fegato dove sono ritrasformati in glucosio. Alcuni tessuti, come gli eritrociti, la midollare del rene e il tessuto adiposo, ottengono di norma l’energia attraverso l’ossidazione parziale del glucosio a piruvato e lattato. Il muscolo scheletrico ricorre alla glicolisi anaerobia in condizioni di ipossia durante l’attività fisica intensa. Nel cervello e nel fegato il glucosio è completamente ossidato ad anidride carbonica ed acqua.
2.1. GLUCOSIO
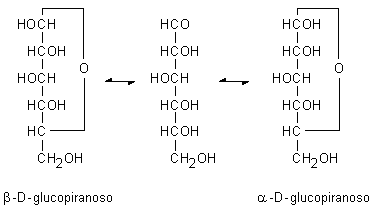
Fig. 2.1. D-glucosio. Il glucosio forma un legame emiacetalico interno stabile fra il gruppo aldeidico in posizione 1 e il gruppo alcolico in posizione 5 e dà luogo a due stereoisomeri, l’α-D-glucosio e il β-D-glucosio, che hanno un nuovo centro di asimmetria sull’atomo di carbonio in 1. La forma a catena aperta del glucosio si trova soltanto in minime quantità nelle soluzioni acquose, ma è un intermedio nell’interconversione delle forme α e β durante la mutarotazione.
In condizioni fisiologiche il livello della glicemia è controllato dall’equilibrio tra produzione e utilizzazione del glucosio. La regolazione della glicemia è il risultato dell’interazione tra insulina e glucagone. L’insulina favorisce la conservazione dei fattori della nutrizione, promuovendo la sintesi del glicogeno e delle proteine e la lipogenesi. La funzione principale del glucagone è invece quella di prevenire l’ipoglicemia attraverso la stimolazione della glicogenolisi e della gluconeogenesi. In seguito ad una consueta assunzione di cibo, l’aumento del glucosio e dell’insulina stimola la glicogeno sintetasi ed inibisce la glicogeno fosforilasi con conseguente netto aumento del glicogeno epatico. Dopo 3-4 ore dal pasto, i livelli di insulina e di glucosio iniziano a scendere e nel fegato torna a prevalere una netta immissione del glucosio in circolo. All’inizio, il 75% della produzione del glucosio epatico è dovuto alla glicogenolisi e il 25% alla gluconeogenesi, ma col ridursi delle scorte di glicogeno nel fegato, il contributo della gluconeogenesi aumenta nettamente nel tempo. Col protrarsi del digiuno, la diminuzione dell’insulina plasmatica è accompagnata da un aumento della concentrazione degli acidi grassi e dei corpi chetonici che possono essere utilizzati come substrati energetici alternativi, limitando la necessità di glucosio.
Se la concentrazione di glucosio si abbassa sotto i valori normali, entra in funzione un meccanismo di emergenza addizionale, costituito dalla increzione di adrenalina. Questa attiva ulteriormente la glicogenolisi e può, a sua volta, stimolare la produzione di ACTH, che porta ad un incremento della concentrazione degli ormoni corticosteroidi e alla attivazione dei processi gliconeogenetici (vedi Par. 10.5.1).
2.1a. Diabete mellito
Il dosaggio della glicemia è di fondamentale importanza sia
per la diagnosi che per il trattamento del diabete mellito
![]() . Questa è una sindrome
eterogenea caratterizzata da iperglicemia e dalla predisposizione
ad andare incontro a complicanze croniche quali la
microangiopatia (responsabile della retinopatia e nefropatia
diabetica) e la macroangiopatia (caratterizzata dal processo
aterosclerotico a rapida evoluzione che colpisce le arterie di
calibro maggiore e medio). Sono state proposte diverse
classificazioni per il diabete. Esse tendono a riunire i pazienti
in gruppi con storia naturale e trattamento simili, tenendo anche
conto delle cause della malattia, quando questo è possibile (Tab. 2.I).
. Questa è una sindrome
eterogenea caratterizzata da iperglicemia e dalla predisposizione
ad andare incontro a complicanze croniche quali la
microangiopatia (responsabile della retinopatia e nefropatia
diabetica) e la macroangiopatia (caratterizzata dal processo
aterosclerotico a rapida evoluzione che colpisce le arterie di
calibro maggiore e medio). Sono state proposte diverse
classificazioni per il diabete. Esse tendono a riunire i pazienti
in gruppi con storia naturale e trattamento simili, tenendo anche
conto delle cause della malattia, quando questo è possibile (Tab. 2.I).
Tab. 2.I. Classificazione del diabete mellito
| Diabete di tipo 1 | |||
| Autoimmune | |||
| Idiopatico | |||
| Diabete di tipo 2 | |||
| Altri tipi specifici di diabete | |||
| Difetti genetici riguardanti la funzione delle cellule beta | |||
| diabete giovanile ad insorgenza tardiva (MODY) | |||
| MODY 1 - deficit del fattore epatico nucleare-4α (HNF-4α) | |||
| MODY 2 - deficit di glucochinasi | |||
| MODY 3 - deficit del fattore epatico nucleare-1α (HNF-1α) | |||
| MODY 4 - deficit del fattore del promotore dell’insulina (IPF-1) | |||
| MODY 5 - deficit del fattore epatico nucleare-1β (HNF-1β) | |||
| MODY 6 - deficit della proteina neurod-1 | |||
| malattie mitocondriali | |||
| MIDD (Maternally Inherited Diabetes with Deafness) | |||
| MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, Stoke-like episodes) | |||
| KSS (Kearns-Sayre syndrome) | |||
| altri difetti genetici riguardanti l’insulina | |||
| Difetti genetici riguardanti la funzione dell’insulina | |||
| resistenza all’insulina tipo A | |||
| leprecaunismo | |||
| sindrome di Rabson-Mendenhall | |||
| Altre sindromi genetiche associate a diabete | |||
| Malattie del pancreas esocrino | |||
| pancreatopatia fibrocalcolotica | |||
| pancreatiti | |||
| traumi accidentali o operatori | |||
| neoplasie | |||
| fibrosi cistica | |||
| emocromatosi | |||
| Malattie endocrine | |||
| sindrome di Cushing | |||
| acromegalia | |||
| ipertiroidismo | |||
| feocromocitoma | |||
| glucagonoma | |||
| somatostatinoma | |||
| Farmaci o agenti chimici | |||
| Infezioni | |||
| Forme di diabete immunomediato | |||
| autoanticorpi anti-insulina | |||
| autoanticorpi anti-recettore per l’insulina (resistenza all’insulina tipo B) | |||
| Stiff-man syndrome | |||
| Diabete gestazionale | |||
|
|||
Il diabete di tipo 1 è dovuto ad una distruzione delle cellule beta del pancreas ed è caratterizzato da una tendenza verso la chetoacidosi. Il più delle volte, il danno è causato da una reazione autoimmune cellulo-mediata per la presenza di anticorpi specifici (anti-isola pancreatica, ICA; anti-insulina, IAA; anti-glutammato decarbossilasi, GAD65A; anti-tirosina fosfatasi, IA-2 e IA-2β). La malattia ha un carattere multigenico (vedi Par. 14.3) in cui ciascun trait agisce aumentando o diminuendo la predisposizione di base. Il polimorfismo dei geni del complesso maggiore di istocompatibilità di classe II condiziona in maniera determinante il rischio per la malattia (circa il 40-50% del rischio genetico è attribuibile ai geni HLA-DRB1, HLA-DQA1 e HLA-DQB1). Il grado di distruzione delle cellule beta è variabile, generalmente rapido nei bambini e nei giovani, più lento negli adulti (in quest’ultimo caso si parla di Latent Autoimmune Diabetes of the Adult, LADA). Altre forme di diabete, più comuni in soggetti di origine africana o asiatica, non hanno invece una etiologia nota e sono caratterizzate da carenza assoluta di insulina e tendenza alla chetoacidosi senza che vi siano segni di autoimmunità o predisposizioni genetiche legate al complesso HLA.
Nel diabete di tipo 2 (definito precedentemente come diabete mellito non insulino-dipendente o diabete a insorgenza nell’adulto) non vi è distruzione autoimmune del pancreas. La chetoacidosi non è frequente e di solito compare in associazione con altre patologie, come ad esempio un’infezione. La maggioranza dei pazienti è obesa e l’obesità in sé è causa o aggrava l’insulino-resistenza. Questa forma di diabete resta frequentemente non diagnosticata per molti anni perché l’iperglicemia non è severa al punto da dare sintomi evidenti. Malgrado ciò, i pazienti affetti da diabete di tipo 2 hanno un rischio elevato di sviluppare complicanze micro- e macroangiopatiche.
Diversi tipi di diabete possono essere associati a difetti monogenici della
funzione beta-cellulare, ereditati di solito in forma autosomica dominante. Queste
alterazioni genetiche sono generalmente caratterizzate dalla comparsa di una modesta
iperglicemia in età relativamente giovane, di solito prima dei 25 anni, e per questo motivo
sono state identificate come diabete giovanile ad insorgenza tarda (Maturity Onset
Diabetes of Youth,
MODY). Tra le forme più frequenti c’è quella associata ad
una
mutazione sul gene della glucochinasi, tale da rendere necessari livelli di
glucosio più elevati per evocare una normale increzione di insulina
![]() . Altre forme
sono legate ad alterazioni di fattori di trascrizione nucleare. Mutazioni
puntiformi del DNA mitocondriale possono essere associate a diabete mellito e
sordità o ad altre sindromi (vedi Par. 14.4). In alcune famiglie sono state identificate anomalie genetiche che comportano
l’incapacità di convertire la proinsulina a insulina oppure
sono state trovate molecole mutanti di insulina con bassa affinità per il
recettore periferico; entrambe queste situazioni portano ad una lieve
intolleranza ai carboidrati.
. Altre forme
sono legate ad alterazioni di fattori di trascrizione nucleare. Mutazioni
puntiformi del DNA mitocondriale possono essere associate a diabete mellito e
sordità o ad altre sindromi (vedi Par. 14.4). In alcune famiglie sono state identificate anomalie genetiche che comportano
l’incapacità di convertire la proinsulina a insulina oppure
sono state trovate molecole mutanti di insulina con bassa affinità per il
recettore periferico; entrambe queste situazioni portano ad una lieve
intolleranza ai carboidrati.
Mutazioni sul gene per il
recettore dell’insulina possono dare luogo a diverse
sindromi la cui gravità dipende dalla presenza dell’alterazione
su uno o entrambi gli alleli; la maggior parte dei casi (resistenza
all’insulina tipo A, leprecaunismo, sindrome di Rabson-Mendenhall)
si accompagna a acanthosis nigricans e, nelle donne, a
iperandrogenismo
![]() . Altre sindromi
genetiche associate a diabete o ad alterata tolleranza al glucosio sono,
ad esempio, la distrofia miotonica
. Altre sindromi
genetiche associate a diabete o ad alterata tolleranza al glucosio sono,
ad esempio, la distrofia miotonica
![]() , la corea di Huntington
, la corea di Huntington
![]() , l’atassia di
Friedreich
, l’atassia di
Friedreich
![]() ,
la sindrome di Prader-Willi
,
la sindrome di Prader-Willi ![]() , la sindrome di Wolfram
, la sindrome di Wolfram
![]() , la sindrome di Werner
, la sindrome di Werner
![]() , la sindrome di Cockayne
, la sindrome di Cockayne
![]() , la sindrome di Down
, la sindrome di Down
![]() , la sindrome di
Klinefelter
, la sindrome di
Klinefelter
![]() e
la sindrome di Turner
e
la sindrome di Turner
![]() .
.
Il diabete mellito può essere secondario a
malattie pancreatiche. Una delle più comuni è
la pancreatite cronica dell’alcolista. Il danno pancreatico può inoltre
essere conseguente a fibrocalcolosi, traumi, neoplasie o ad alcune alterazioni
metaboliche (fibrosi cistica
![]() , emocromatosi
, emocromatosi
![]() ).
).
Il diabete può inoltre essere causato da una iperincrezione di ormoni ad effetto anti-insulinico, come in caso di sindrome Cushing (vedi Par. 10.5.1d), acromegalia (vedi Par. 10.2), ipertiroidismo (vedi Par. 10.4), feocromocitoma, glucagonoma o somatostatinoma. Molti farmaci determinano alterazioni della increzione insulinica; generalmente questi farmaci non causano di per sé il diabete, ma possono precipitarlo in persone con insulino-resistenza. I farmaci diuretici possono causare diabete inducendo un iperaldosteronismo secondario (vedi Par. 10.5.1d) e una deplezione di K+ , che interferisce a sua volta con la increzione di insulina. Fra le cause infettive di diabete bisogna ricordare la rosolia congenita e l’infezione da citomegalovirus.
Il diabete può essere associato a diverse patologie immunologiche, con patogenesi ed etiologia differenti da quelle che conducono al diabete tipo 1. Una iperglicemia postprandiale di entità sufficiente a soddisfare i criteri diagnostici per il diabete è stata osservata in quei rari soggetti che sviluppano spontaneamente anticorpi anti-insulina, anche se, in realtà, i pazienti presentano più spesso sintomi di ipoglicemia, piuttosto che di iperglicemia (vedi Tab. 2.II). Gli anticorpi anti-recettore per l’insulina si ritrovano occasionalmente in pazienti con lupus eritematoso o altre patologie autoimmuni (questi anticorpi possono causare diabete, quando ostacolano il legame dell’insulina ai tessuti bersaglio, o ipoglicemia, quando agiscono da agonisti insulinici); come in altre condizioni di estrema insulino-resistenza, i pazienti con anticorpi anti-recettore per l’insulina hanno spesso acanthosis nigricans. La Stiff-man syndrome è un disordine autoimmune del sistema nervoso centrale caratterizzato da rigidità muscolare e spasmi dolorosi; i pazienti presentano solitamente alti titoli di anticorpi anti GAD e, nella metà dei casi, sviluppano una forma di diabete.
Il termine "diabete gestazionale" si riferisce a qualsiasi grado di intolleranza al glucosio che esordisce o viene diagnosticato durante la gravidanza, più frequentemente durante il terzo trimestre. Molto spesso si tratta di un tipico diabete mellito di tipo 2 che viene smascherato dallo stress della gravidanza. Le pazienti generalmente presentano una ridotta increzione di insulina e, nei 3/4 dei casi, una anamnesi familiare positiva per il diabete. Il riconoscimento clinico del diabete gestazionale è importante perché la sorveglianza e l’intervento sanitario possono ridurre la morbosità e la mortalità neonatale. L’accertamento dei fattori di rischio (obesità, familiarità per il diabete, glicosuria, pregresso diabete gestazionale) deve essere effettuato all’inizio della gravidanza.
2.1b. Ipoglicemie
I disordini che sono causa di un abbassamento della concentrazione di glucosio nel sangue sono classificati in ipoglicemie reattive e ipoglicemie a digiuno, in base al tempo di comparsa dei sintomi in relazione ai pasti. Nelle ipoglicemie reattive (o postprandiali) la sintomatologia non si presenta mai a digiuno, ma compare tipicamente dopo 2-5 ore da un pasto ed è di tipo prevalentemente adrenergico. Nelle ipoglicemie a digiuno i sintomi sono esacerbati dall’attività fisica e compaiono solitamente dopo un periodo prolungato di digiuno (ad esempio prima della colazione del mattino) o quando si omette di consumare uno dei pasti. Le ipoglicemie a digiuno si distinguono in ipoglicemie da mancata produzione di glucosio ed ipoglicemie da sovrautilizzazione di glucosio. Ulteriori sottoclassificazioni sono riportate in Tab. 2.II.
Tab. 2.II. Classificazione delle ipoglicemie
| Ipoglicemie reattive | |||
| idiopatica | |||
| iperinsulinismo alimentare | |||
| intolleranza ereditaria al fruttosio | |||
| galattosemia | |||
| Ipoglicemie a digiuno | |||
| da mancata produzione di glucosio | |||
| difetti ormonali | |||
| ipopituitarismo | |||
| insufficienza surrenalica | |||
| deficit di catecolamine | |||
| deficit di glucagone | |||
| difetti enzimatici | |||
| glucosio 6-fosfatasi | |||
| fosforilasi epatica | |||
| piruvato carbossilasi | |||
| fosfoenolpiruvato carbossichinasi | |||
| fruttosio 1,6-difosfatasi | |||
| glicogeno sintetasi | |||
| deficit di substrato | |||
| malnutrizione | |||
| gravidanza avanzata | |||
| malattie epatiche acquisite | |||
| farmaci | |||
| da sovrautilizzazione di glucosio | |||
| iperinsulinismo | |||
| insulinoma | |||
| insulina esogena | |||
| sulfonilurea | |||
| anticorpi anti-insulina o anti-recettore per l’insulina | |||
| farmaci (chinino, etc.) | |||
| shock da endotossine | |||
| altro | |||
| tumori extrapancreatici | |||
2.1.1. Metodi di determinazione
I più antichi metodi determinazione del glucosio erano basati sulla riduzione degli ioni rameici bivalenti (Cu++) in ioni rameosi monovalenti (Cu+). Questi ultimi formano a caldo dell’ossido rameoso (Cu2O) che può essere messo in evidenza in vari modi. Il più comune è la riduzione del fosfomolibdato (nella reazione di Folin-Wu) o dell’arsenomolibdato (nella reazione di Somogyi-Nelson) in blu di molibdeno. La reazione secondo Benedict, che prevede l’evidenziazione diretta dell’ossido rameoso di colore rosso, è ancora in uso nel dosaggio semiquantitativo del glucosio nelle urine. Sovradosaggi dell’analita possono essere dovuti ad altre sostanze riducenti (vitamina C, creatinina, urato) presenti nel campione. Il metodo, in combinazione con i test enzimatici altamente specifici per il glucosio, permette inoltre di determinare la presenza di altri zuccheri riducenti nelle urine di neonati con malattie ereditarie riguardanti il metabolismo dei carboidrati. Un test enzimatico negativo associato ad una reazione di Benedict positiva è suggestivo per una tale eventualità (vedi Par. 14.6.4a).
Altri metodi non enzimatici per la determinazione del glucosio (attualmente usati molto di rado) prevedono la riduzione in ambiente alcalino del ferricianuro giallo, Fe(CN)6-3, in ferrocianuro incolore, Fe(CN)6-4. Quest’ultimo può reagire a sua volta con gli ioni ferrici per formare ferrocianuro ferrico (blu di Prussia). La reazione all’o-toluidina è invece basata sulla capacità di questa amina aromatica (da adoperarsi con cautela perché potenzialmente cancerogena) di condensare in ambiente acido con il gruppo aldeidico del glucosio per formare una glicosamina (Fig. 2.2).
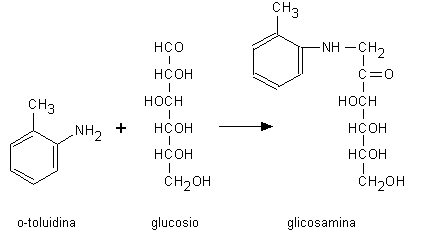
Fig. 2.2. La reazione dell’o-toluidina per il dosaggio del glucosio
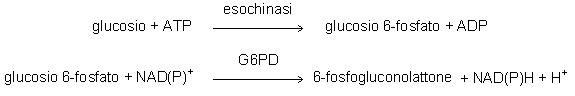
I metodi più frequentemente usati oggigiorno sono enzimatici e si basano sulle reazioni catalizzate dalla esochinasi, dalla glucosio ossidasi o dalla glucosio deidrogenasi. I metodi che si basano sull’esochinasi prevedono di accoppiare a questo enzima la glucosio 6-fosfato deidrogenasi (G6PD) e di osservare la reazione seguendo la riduzione del NAD(P)+ in NAD(P)H. L’uso di una glucosio 6-fosfato deidrogenasi batterica, capace di adoperare il NAD+ al posto del NADP+, evita eventuali interferenze da parte dell’enzima umano presente nei globuli rossi che è specifico solo per NADP+. L’esochinasi può fosforilare anche altri esosi, ma questi sono generalmente presenti a concentrazioni molto basse e non causano significative interferenze.
La glucosio ossidasi ossida il glucosio a gluconolattone ed acqua ossigenata. Poiché l’enzima è altamente specifico per il β-D-glucosio, molti prodotti commerciali contengono anche della mutarotasi che catalizza la conversione dell’a-D-glucosio nella forma b (vedi Fig. 2.1). La reazione catalizzata dalla glucosio ossidasi può essere seguita per mezzo di un elettrodo ad ossigeno o aggiungendo della perossidasi e uno dei numerosi substrati di questo enzima (4-aminofenazone, 3-metil-2-benzotiazolinone idrazone/N,N-dimetilanilina, 3,3’,5,5’-tetrametilbenzidina, 1,7-diidrossinaftalene, KI) che danno luogo a prodotti colorati. La procedura è applicabile ad apparecchi automatizzati o a strisce reattive per una determinazione estemporanea della glicemia. Una ulteriore variante del metodo prevede di utilizzare, come substrati della perossidasi, il 4-aminoantipirene e l’acido sulfonico del 3,5-dicloro-2-idrossibenzene: la reazione è seguita non misurando l’assorbimento del cromogeno, ma la capacità del prodotto di reazione di attenuare la fluorescenza della fluoresceina presente nella miscela (radiation energy attenuation).

La glucosio deidrogenasi catalizza la ossidazione del glucosio in gluconolattone. La reazione può essere facilmente seguita seguendo la formazione di NADH a 340 nm.
![]()
2.1.2. Preparazione del campione ed intervalli di riferimento
Il glucosio è dosato preferibilmente nel siero o nel plasma privi di emolisi e, più raramente, in altri liquidi biologici, come il fluido cerebrospinale e l’urina. I comuni anticoagulanti (ossalato, fluoruro, EDTA, citrato, eparina) non causano interferenze con l’analisi. Il campione di sangue deve essere centrifugato al più presto e la parte corpuscolata deve essere rimossa in quanto nel sangue intero a temperatura ambiente il glucosio viene degradato attraverso la via glicolitica ad una velocità di circa il 5% all’ora. Se non è possibile centrifugare subito il campione, è necessario aggiungere ad esso degli inibitori della glicolisi, come il fluoruro o lo iodoacetato. Quest’ultimo composto è usato più frequentemente rispetto al fluoruro perché, in un secondo tempo, permette una più facile preparazione del siero. Il glucosio nel plasma o nel siero privi di cellule è stabile fino a 3 giorni a 2°- 8°C.
La determinazione del glucosio nell’urina è di costo limitato, ma pone diversi problemi interpretativi. L’impiego di questo test sta diminuendo nei paesi occidentali, anche se nei pazienti anziani con diabete mellito non insulino-dipendente, trattati solamente con dieta o con piccole dosi di ipoglicemizzanti orali, può essere più accettabile del dosaggio nel sangue e fornire una affidabile indicazione del controllo glicemico.
Storicamente, vi è stata divergenza di opinione circa la dose di glucosio da impiegare nel test di tolleranza al glucosio per via orale. Le attuali raccomandazioni dell’Organizzazione Mondiale di Sanità consigliano di somministrare una soluzione contenente 1,75 g di glucosio anidro per kg di peso corporeo fino ad un massimo di 75 - 100 g. Nei tre giorni precedenti il test è consentita al paziente una dieta libera (con almeno 200 g di carboidrati al giorno) e una normale attività fisica. Il paziente deve osservare il digiuno la notte precedente il test per un periodo di almeno 8-10 ore e, durante la prova, deve rimanere seduto senza fumare. Il test è sconsigliabile ai pazienti che presentino iperglicemia a digiuno e a quelli ospedalizzati con affezioni acute o immobilizzati. L’interpretazione del test può presentare difficoltà nei pazienti trattati con β-bloccanti, diuretici, acido nicotinico o alte dosi di alcuni ormoni.
Nei soggetti sani la concentrazione del glucosio ematico è mantenuta entro limiti assai poco variabili da un soggetto all’altro. Nel periodo postassorbimento (ad esempio dopo una notte a digiuno) la glicemia è compresa tra 4,5 e 5,2 mmol/L con coefficienti di variazione intraindividuali dell’1-2%. I coefficienti di variazione interindividuali sono inferiori al 5%, a condizione che l’intervallo di tempo dall’ultimo pasto, la sua composizione e l’entità dell’attività fisica siano simili. Nei soggetti sani, la glicemia dopo un pasto consueto non supera le 10 mmol/L e ritorna nei limiti normali entro 2-4 ore. Un abbassamento della glicemia è un evento inconsueto nelle ordinarie attività dell’uomo adulto nei paesi occidentali. In genere, si considera ipoglicemia una concentrazione di glucosio venoso inferiore a 2,5 mmol/L, indipendentemente dalla presenza o meno della sintomatologia.
La diagnosi di diabete si basa o sul livello glicemico a digiuno o sulla
risposta ad un carico orale di glucosio. Secondo le attuali direttive, per fare
diagnosi di diabete mellito è necessario che sia soddisfatto almeno uno dei
seguenti criteri
![]() :
:
1. Presenza di sintomi riferibili al diabete con un valore di glicemia
misurata a caso uguale o superiore a 200 mg/dL (11.1 mmol/L). I sintomi
riferibili al diabete sono poliuria, polidipsia e/o una perdita di peso non
spiegabile altrimenti. La misura della glicemia viene effettuata in un qualsiasi
momento della giornata, indipendentemente dai pasti.
2. Glicemia a digiuno uguale o superiore a 126 mg/dL (7,0 mmol/L). Per
digiuno si intende la mancata introduzione di cibo da almeno 8 ore.
3. Glicemia uguale o superiore a 200 mg/dL (11.1 mmol/L) dopo 2 o più ore da un carico
orale contenente l’equivalente di 75 g di glucosio anidro.
Tutte le donne gravide dovrebbero essere esaminate tra la ventiquattresima e la ventottesima settimana di gravidanza con un carico orale di 50 g di glucosio, seguito un’ora dopo dalla determinazione della glicemia. Il test di screening per il diabete gestazionale può essere effettuato in qualunque momento della giornata indipendentemente dai pasti. Se la glicemia postprandiale a un’ora è maggiore o uguale a 140 mg/dL, si deve eseguire un test da carico di glucosio da 100 g.
2.2. GLICAZIONE NON ENZIMATICA DELLE PROTEINE
Le proteine possono reagire spontaneamente con il glucosio o
altri monosaccaridi (glucosio 6-fosfato, galattosio, acido
sialico, pentosi) dando luogo a una serie di prodotti di
condensazione
![]() .
La prima reazione nel processo di glicazione consiste nella
formazione di una base di Schiff fra il gruppo riducente dello
zucchero e un gruppo aminico della proteina (gruppo aminico N-terminale,
gruppo e-aminico di una lisina, gruppo
aminico della porzione guanidinica di una arginina). La reazione
è reversibile e procede rapidamente. La base di Schiff va poi
incontro a un lento riarrangiamento intramolecolare con
formazione di un prodotto stabile (prodotto di Amadori) che tende
ad accumularsi (Fig. 2.3).
.
La prima reazione nel processo di glicazione consiste nella
formazione di una base di Schiff fra il gruppo riducente dello
zucchero e un gruppo aminico della proteina (gruppo aminico N-terminale,
gruppo e-aminico di una lisina, gruppo
aminico della porzione guanidinica di una arginina). La reazione
è reversibile e procede rapidamente. La base di Schiff va poi
incontro a un lento riarrangiamento intramolecolare con
formazione di un prodotto stabile (prodotto di Amadori) che tende
ad accumularsi (Fig. 2.3).
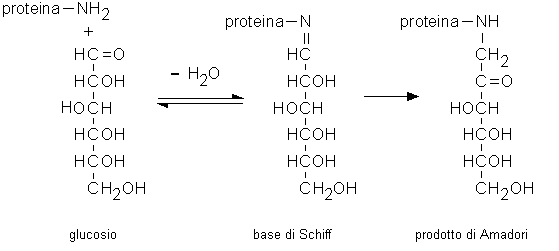
Fig. 2.3. Glicazione non enzimatica delle proteine
Il prodotto di Amadori si modifica ulteriormente e genera, sempre lentamente, composti di glicazione colorati (AGE, advanced glycation end-products) che formano legami crociati con proteine o altre macromolecole contenenti gruppi aminici liberi. Un AGE isolabile dai tessuti è ad esempio il 2-furoil-4-(5)-(2-furanil)-1H imidazolo (FFI) che si forma a partire da glucosio e coinvolge due residui di lisina della proteina (Fig. 2.4).
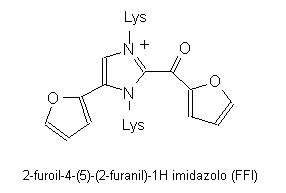
Fig. 2.4. AGE (advanced glycation end-products)
Il grado di glicazione di una proteina dipende da vari fattori, quali la concentrazione e le caratteristiche fisico-chimiche del saccaride e della proteina, il tempo di interazione, il pH del mezzo (la reazione di Amadori non avviene a pH inferiore a 7). Nell’anziano, dove col tempo i processi di glicazione raggiungono uno stadio avanzato, i tessuti ricchi di proteine longeve (collagene, proteine del cristallino, membrana basale glomerulare) si arricchiscono di composti AGE, esibendo così una maggiore rete di legami crociati. Molti processi fisiologici risultano alterati a seguito della glicazione delle proteine: numerosi enzimi (catepsina B, ribonucleasi pancreatica, esosaminidasi) subiscono una forte diminuzione di attività; l’emoglobina e l’antitrombina III riducono la loro affinità di legame; le lipoproteine plasmatiche interagiscono meno facilmente con i recettori specifici; l’albumina e le proteine della mielina sono assunte più facilmente rispettivamente dalle cellule endoteliali e dai macrofagi.
I fenomeni di glicazione diventano particolarmente importanti sul piano sia patogenetico che diagnostico nella sindrome diabetica. In tali circostanze aumenta infatti la glicazione delle proteine plasmatiche in generale e dell’albumina in particolare. Inoltre aumenta la glicazione dell’emoglobina. L’emoglobina A1 è la frazione glicoproteica più comunemente dosata nei soggetti diabetici (vedi Par. 7.2). Essa è a sua volta separabile in diverse subfrazioni (Tab. 2.III), fra le quali la più importante è l’Hb A1c.
Tab. 2.III - Emoglobine glicate
| Nome | catena interessata | monosaccaride |
| HbA1a1 | catena β: gruppo aminico N-terminale | fruttosio 1,6-difosfato |
| HbA1a2 | catena β: gruppo aminico N-terminale | glucosio 6 fosfato |
| HbA1b | non ancora definita in dettaglio |
|
| HbA1c | catena β: gruppo aminico N-terminale | glucosio |
| HbA1d | catene α e β: gruppi
aminici in ε
della lisina e gruppo aminico N-terminale |
|
2.2.1. Metodi di determinazione
La determinazione complessiva delle diverse frazioni della emoglobina glicata può essere ottenuta mediante metodi cromatografici o elettroforetici. Nel primo caso si sfrutta la minore affinità dell’emoglobina glicata per le resine a scambio cationico. Nel secondo caso si sfrutta il fatto che il destran solfato o altri composti simili si legano alla emoglobina nativa cambiandone la mobilità elettroforetica, ma non interferiscono sulla corsa elettroforetica della emoglobina glicata. Un altro approccio, che non prevede la separazione dell’analita, si basa sulla trasformazione del complesso di Amadori nella base di Schiff del 5-idrossimetilfurfurale, nella sua idrolisi dalla proteina e nella sua reazione con l’acido 2-tiobarbiturico (Fig. 2.5).
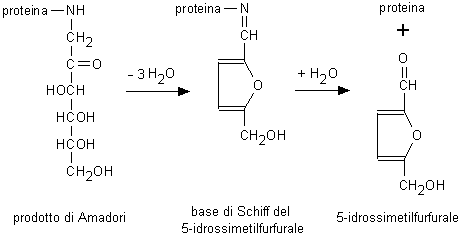
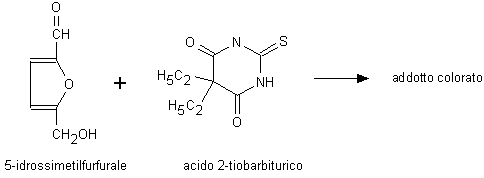
Fig. 2.5. Determinazione del complesso di Amadori con l’acido 2-tiobarbiturico
La determinazione della frazione HbA1c può essere ottenuta mediante cromatografia liquida ad alta pressione (HPLC), cromatografia per affinità o elettrofocalizzazione al punto isoelettrico. Un ulteriore metodo sfrutta la capacità dell’inositolo esafosfato di legarsi in prossimità della regione N-terminale della catena β , cambiando lo spettro di assorbimento dell’emoglobina, e la capacità del glucosio presente sul gruppo N-terminale nella HbA1c di impedire lo stabilirsi di questo legame.
La presenza di HbF può determinare una sovrastima dell’emoglobina glicata nella maggior parte dei casi, salvo quando si usa la cromatografia per affinità o il metodo all’inositolo esafosfato. Le resine a scambio cationico possono sottostimare l’emoglobina glicata nel caso di HbS e HbC o sovrastimarla quando sono presenti dei lipidi. Il metodo dà inoltre risultati errati quando il gruppo N-terminale è modificato da salicilati, carbamato o galattosio.
Il metodo che frutta la reazione dell’acido 2-tiobarbiturico con il 5-idrossimetilfurfurale e quello basato sulla separazione cromatografica per affinità sono usati anche nella determinazione dell’albumina e delle proteine totali glicate. Per queste frazioni proteiche è anche usato un metodo (saggio della fruttosamina) che sfrutta la capacità riducente del gruppo chetoaminico del complesso di Amadori sul nitroblu di tetrazolio.
2.2.2. Preparazione del campione ed intervalli di riferimento
Il sangue deve essere raccolto in EDTA e la parte corpuscolata deve essere separata dal plasma. Per la determinazione delle emoglobine glicate, gli eritrociti devono essere lavati una volta in 0,15 M NaCl e risospesi nel medesimo mezzo in un volume pari a quello del campione originale di sangue. Le cellule lavate o l’emolizzato possono essere conservati fino ad una settimana a 4°C e fino ad un mese a –70°C. La quantità di emoglobina glicata "labile" è generalmente bassa. Si è visto infatti che la quantità di HbA1 aumenta solo del 1-2% (rispetto all’emoglobina totale) dopo un test di tolleranza al glucosio per via orale o dopo aver incubato le emazie lavate in un mezzo contenente 500 mg/L di glucosio. Ugualmente si sono osservate solo piccole diminuzioni nel livello di HbA1 dopo aver dializzato il campione. Comunque, se si desidera eliminare la forma labile, è consigliabile dializzare le emazie in 0,15 M NaCl o incubarle brevemente in presenza di semicarbizide e anilina a pH 5.
I valori di riferimento per l’emoglobina glicata variano con il variare della metodica impiegata. In generale, in soggetti non diabetici la concentrazione percentuale delle emoglobine glicate varia tra il 6% e l’8%, ma può raggiungere valori del 15-18% in diabetici con uno scarso controllo della propria glicemia. La concentrazione percentuale di HbA1c nei soggetti non diabetici è compresa tra il 4% e il 5%.
Per quanto riguarda l’albumina glicata, non si trovano dei costanti valori di riferimento nei fogli illustrativi che accompagnano i diversi prodotti commerciali utilizzati per l’analisi. Questi sono compresi tra l’1,2% e il 2,3% in alcuni casi e tra l’8,4% e il 15,7% in altri.
2.3. Acido lattico
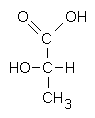
Fig. 2.6. Acido lattico
L’acido lattico si è forma nelle cellule muscolari per mantenere un adeguato livello di NAD+. Infatti, quando l’apporto di ossigeno è diventa insufficiente, la cellula deve dipendere sempre più dalla glicolisi anaerobia per formare l’ATP. Per rigenerare l’NAD+ necessario per metabolizzare il glucosio attraverso questa via, la cellula deve ossidare l’NADH convertendo in acido lattico l’acido piruvico, che è il prodotto terminale della glicolisi stessa. L’acido lattico muscolare, che si accumula durante l’anossia tessutale, viene rilasciato in circolo e metabolizzato in gran parte dal cuore e dal fegato.
Un vigoroso esercizio fisico accompagnato ad ipossia può dar luogo ad una lieve acidosi per accumulo di acido lattico, che è accompagnata a dolori e crampi muscolari e che scompare appena l’attività cessa. Una acidosi per accumulo di acido lattico più grave si può avere quando l’ipossia è causata da una insufficienza respiratoria, da uno stato di ipoperfusione (ad esempio, nel collasso circolatorio) o da un severo stato di deidratazione con ridotto apporto di ossigeno ai tessuti (ad esempio, nella chetoacidosi diabetica). Anche condizioni che portano ad un aumento del consumo di ossigeno (sepsi, tumori maligni) possono dar luogo ad acidosi lattica.
2.3.1. Metodi di determinazione
L’acido lattico può essere ossidato ad acetaldeide in presenza di permanganato o diossido di manganese con produzione di CO2 o di CO. L’acetaldeide è a sua volta misurata colorimetricamente o mediante gas-cromatografia, mentre il monossido e il diossido di carbonio sono determinati per via gasometrica. I metodi sono accurati, precisi e sensibili, ma richiedono lunghi tempi per l’analisi.
I metodi enzimatici utilizzano la lattico deidrogenasi per ossidare l’acido lattico ad acido piruvico, riducendo l’NAD+ in NADH.
![]()
Alla miscela devono essere aggiunte dell’idrazina o della semicarbazide per rimuovere l’acido piruvico prodotto e portare così a compimento la reazione, che altrimenti risulterebbe fortemente spostata a sinistra. In alternativa ci si può avvalere dell’ossidazione dell’acido lattico ad opera del ferricianuro. La reazione è catalizzata da una deidrogenasi di lievito. Il ferrocianuro che si forma viene a sua volta misurato elettrochimicamente con un elettrodo di platino contro Ag/AgCl.
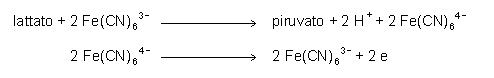
Un altro metodo prevede l’utilizzo della lattato ossidasi e la misura elettrochimica del perossido d’idrogeno prodotto.
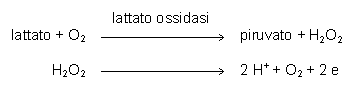
2.3.2. Preparazione del campione ed intervalli di riferimento
Il campione di sangue deve essere prelevato avendo cura di non indurre stasi venosa. A tal fine è conveniente togliere il laccio dopo aver infilato l’ago e attendere qualche minuto prima di aspirare il sangue. Bisogna inoltre evitare che il glucosio presente nel campione venga metabolizzato dalle cellule ad acido lattico, causando una sovrastima dell’analita. Sia il fluoruro di sodio che lo iodoacetato inibiscono efficacemente la glicolisi. Tuttavia il fluoruro di sodio può inibire parzialmente anche la coagulazione e contaminare così il siero con frustoli di fibrina. Lo iodoacetato (0,5 g/L) non disturba invece la normale formazione del coagulo. Nel caso che si usi sangue eparinizzato, è necessario mettere il campione in ghiaccio e separare rapidamente il plasma dagli elementi corpuscolati.
La concentrazione di acido lattico nel sangue venoso è inferiore a 2 mmol/L in un soggetto a digiuno ed a riposo, mentre nel sangue arterioso il livello è la metà o i due terzi di quello venoso. Se il soggetto non è a riposo, la concentrazione di acido lattico è più alta, così come dopo un pasto. Un aumento del livello di acido lattico nel sangue (> 10,5 mmol/L) è un indice prognostico negativo per l’evoluzione del quadro morboso che sta alla base della variazione del quadro ematochimico.
La concentrazione di acido lattico nel fluido cerebrospinale varia da 1,1 a 2,8 mmol/L. Concentrazioni più elevate (> 4 mmol/L) possono riscontrarsi quando vi sono dei tumori che impediscono l’apporto di sangue al tessuto nervoso o in presenza di infezioni batteriche o da miceti, ma non in presenza di infezioni virali.
2.4. ACIDO PIRUVICO
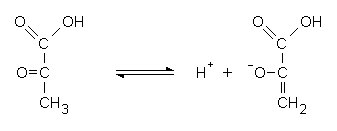
Fig. 2.7. Conversione cheto-enolica dell’acido piruvico. La forma chetonica è favorita a pH acido.
In condizioni di anossia, quando il rapporto NAD/NADH è basso, l’acido piruvico è ridotto ad acido lattico per permettere il metabolismo anaerobio del glucosio. Perciò, durante un esercizio leggero aumentano sia l’acido piruvico che l’acido lattico, mentre in condizioni di anossia il rapporto lattato/piruvato cresce. Questo rapporto può quindi servire per stimare la gravità di una insufficienza di circolo: più alto è il suo valore, più grave è l’anossia.
2.4.1. Metodi di determinazione
L’acido piruvico può essere determinato chimicamente su campioni deproteinizzati in presenza di 2,4-dinitrofenilidrazina. Per eliminare composti interferenti, l’idrazone dell’acido piruvico è estratto in solvente organico (etilacetato, xilene, toluene o benzene) e riportato in ambiente acquoso per aggiunta di Na2CO3. In alternativa il prodotto di reazione è purificato per cromatografia. Tuttavia, il metodo, che prevede una misura fotometrica del prodotto, è poco specifico in quanto altri chetoni endogeni (acido acetacetico, acido levulinico, acido α-chetoglutarico) possono essere responsabili anche del 70% del colore sviluppato, né la cromatografia dei prodotti di reazione semplifica l’interpretazione del dato analitico in quanto per ogni chetone si formano due isomeri dello stesso idrazone.
Il metodo enzimatico si basa sulla riduzione dell’acido piruvico ad acido lattico che può essere seguita spettrofotometricamente a 340 nm. A pH 7,4 la reazione è fortemente spostata verso la formazione di acido lattico.
![]()
2.4.2. Preparazione del campione ed intervalli di riferimento
Il sangue deve essere prelevato da un paziente a digiuno e trattato con iodoacetato per bloccare la glicolisi delle cellule ematiche. Un lieve esercizio fisico o una stasi venosa inferiore a 2 minuti non cambiano i livelli plasmatici di acido piruvico. Il campione deve essere mantenuto al freddo e deproteinizzato appena possibile a causa dell’instabilità dell’analita a temperatura ambiente. I valori di riferimento sono inferiori a 100 m mol/L.
| aggiornamento: 22/02/12 |