|
|
Costantino Salerno |
|
|
Appunti di Biochimica Clinica |
|
|
4. METABOLISMO DEI NUCLEOTIDI E DELLE BASI AZOTATE
La sintesi dei nucleotidi avviene in modo diverso a seconda che essi contengano basi puriniche o basi pirimidiniche. Una caratteristica in comune alle due vie è quella di essere in relazione con lo shunt dei pentosi (o dell’esoso monofosfato). Attraverso lo shunt il glucosio 6-fosfato è ossidato e decarbossilato a ribulosio 5-fosfato e quindi convertito a ribosio 5-fosfato. L’ossidazione diretta del glucosio è particolarmente attiva nei globuli rossi e negli organi e tessuti deputati alla sintesi di acidi grassi, steroidi ed aminoacidi (attraverso la glutammico deidrogenasi): fegato, tessuto adiposo, corteccia surrenalica, testicoli, tiroide e ghiandola mammaria durante l’allattamento. Il ribosio 5-fosfato viene trasformato in 5-fosforibosil-1-pirofosfato (PRPP) attraverso una sintetasi specifica che utilizza l’ATP come donatore di pirofosfato (Fig. 4.1).
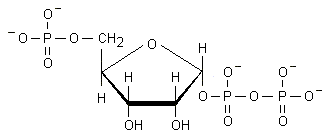
Fig. 4.1. PRPP (5-fosforibosil-1-pirofosfato)
4.1. METABOLISMO PURINICO
I nucleotidi purinici sono sintetizzati sia a partire da precursori più semplici attraverso la “biosintesi de novo”, sia mediante il riutilizzo delle basi azotate e dei nucleosidi attraverso le “vie di recupero” (Fig. 4.2). L’IMP gioca un ruolo centrale nel metabolismo purinico in quanto è, da un lato, il prodotto terminale della biosintesi de novo e, dall’altro lato, un intermedio metabolico necessario alla sintesi dell’AMP e del GMP, i nucleotidi precursori degli acidi nucleici e di numerosi cofattori e secondi messaggeri cellulari. Un altro substrato chiave è il PRPP che è coinvolto sia nelle prime tappe della biosintesi de novo, sia nelle vie di recupero delle basi puriniche.
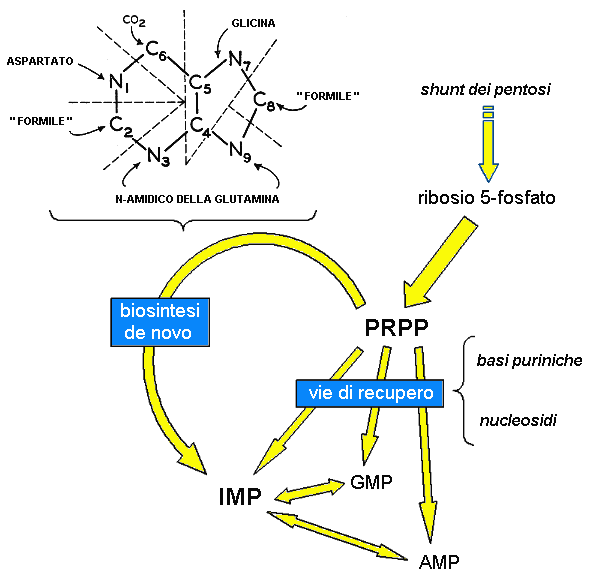
Fig. 4.2. Rappresentazione schematica del metabolismo purinico
4.1a. Biosintesi purinica de novo
La biosintesi de novo delle purine avviene principalmente nel fegato. Altri tessuti, nei quali la biosintesi de novo è meno rappresentata (cervello) o del tutto assente (emazie), dipendono dal fegato per il proprio fabbisogno di purine e si riforniscono di nucleotidi purinici attraverso le vie di recupero. Il globulo rosso svolge un ruolo importante in questo processo in quanto incorpora le purine nel fegato e le ricede ai tessuti che ne abbisognano.
L’IMP è formato attraverso una sequenza lineare di 10 reazioni enzimatiche che portano alla costruzione dell’anello purinico a partire dal PRPP mediante aggiunte successive di atomi di carbonio o di azoto provenienti da aminoacidi (aspartato, glicina, glutamina), anidride carbonica e un derivato formilato dell’acido folico. La via metabolica è regolata attraverso un controllo retroattivo operato dai nucleotidi purinici sulle attività della PRPP sintetasi (che forma PRPP a partire dal ribosio 5-fosfato e dall’ATP) e della glutamina-PRPP amidotransferasi (che forma la 5-fosforibosilamina a spese del PRPP e della glutamina).
4.1b. Vie di interconversione, recupero e degradazione delle purine
Le vie di interconversione, recupero e degradazione delle purine sono fortemente interconnesse (Fig. 4.3). Le vie di interconversione dei nucleotidi coinvolgono gli enzimi inositato deidrogenasi, guanilato sintetasi, guanilato reduttasi, adenilosuccinato sintetasi, adenilosuccinato liasi ed adenilato deaminasi. Le attività di questi enzimi sono soggette a controlli reciproci. Così il GTP è necessario per la conversione dell’IMP in AMP, mentre l’ATP è necessario per la conversione dell’IMP in GMP. Un eccesso di GMP nella cellula inibisce la formazione di XMP dall’IMP ad opera della IMP deidrogenasi, senza alterare la velocità di formazione dell’AMP. Al contrario un accumulo di AMP porta all’inibizione della adenilosuccinato sintetasi, senza modificare la biosintesi del GMP. Il ciclo futile dei nucleotidi purinici (che coinvolge gli enzimi adenilosuccinato sintetasi, adenilosuccinato liasi ed adenilato deaminasi) è operativo specialmente a livello del tessuto muscolare e nervoso e porta alla deaminazione dell’aspartato ed alla produzione di fumarato ed ammoniaca.
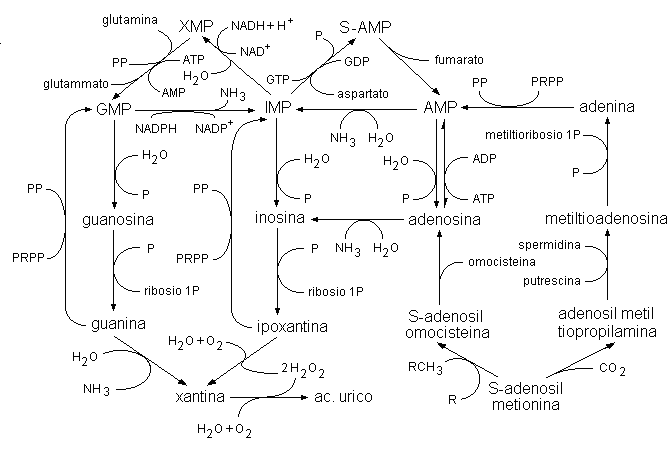
Fig. 4.3. Vie di interconversione purinica
Il recupero dell’ipoxantina e della guanina avviene a spese del PRPP attraverso le due reazioni catalizzate della ipoxantina fosforibosiltransferasi che portano rispettivamente alla formazione di nuovo IMP e nuovo GMP. L’adenina va incontro ad una reazione analoga catalizzata da un enzima specifico (adenina fosforibosiltransferasi), che è ubiquitario nell’organismo ed impedisce che l’adenina possa essere degradata dalla xantina ossidasi a 2,8-diidrossiadenina, un composto estremamente poco solubile e responsabile di gravi danni renali.
Poiché le chinasi specifiche per l’inosina e la guanosina sono assenti nei tessuti umani, il recupero di questi nucleosidi può avvenire solo attraverso la fosforolisi del legame N-glicosidico e la fosforibosilazione delle basi libere per mezzo della ipoxantina fosforibosiltransferasi. L’adenosina, al contrario, può essere fosforilata ad AMP dalla adenosina chinasi a spese dell’ATP. La defosforilazione dell’AMP ad adenosina ad opera della nucleotidasi e la rifosforilazione dell’adenosina ad AMP costituiscono il ciclo futile dell’adenosina che ha lo scopo di regolare i livelli dei nucleotidi adenilici e la disponibilità di adenosina nella cellula.
La reazione catalizzata dall’adenilato deaminasi sembra essere la tappa metabolica cineticamente limitante la degradazione intraepatica dei nucleotidi adenilici ad acido urico, che procede principalmente attraverso la formazione di inositato, inosina, ipoxantina e xantina. L’enzima ha complesse proprietà allosteriche, è inibito dal fosfato inorganico e dal GTP ed è attivato dall’ATP. Si ritiene che, in condizioni fisiologiche, l’adenilato deaminasi sia inibita per circa il 95% dai suoi substrati ed effettori.
La nucleoside fosforilasi purinica catalizza la fosforolisi dell’inosina ad ipoxantina e della guanosina a guanina con formazione di ribosio 1-fosfato, ma non riconosce l’adenosina come substrato. L’adenosina deve essere perciò trasformata in inosina dalla adenosina deaminasi per essere degradata. L’adenina endogena non deriva dall’adenosina, ma è un sottoprodotto nella biosintesi delle poliamine lungo la via che dalla S-adenosilmetionina porta alla formazione della spermidina. I metaboliti dell’adenina endogena costituiscono il 20-30% delle purine escrete.
L’ossidazione della guanina e dell’ipoxantina ad acido urico avviene attraverso le reazioni catalizzate dalla guanasi e dalla xantina ossidasi. Quest’ultimo enzima, che catalizza l’ossidazione sia dell’ipoxantina a xantina sia della xantina ad acido urico, è presente con una elevata attività solo nel fegato e nella mucosa del tenue.
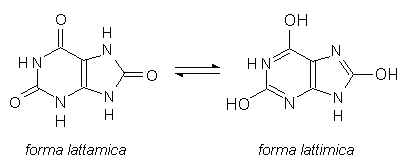
Fig. 4.4. Acido urico. La forma lattimica è debolmente acida (pK’ = 4,5 - 10,3) e forma dei sali scarsamente solubili in acqua.
L’acido urico è il principale catabolita purinico eliminato con le urine. L’acido urico è filtrato attraverso i glomeruli (ad eccezione forse di una piccola quota legata alle proteine), riassorbito in gran parte e secreto nuovamente dai tubuli probabilmente attraverso lo stesso meccanismo responsabile per la secrezione di altri anioni organici.
Sono note le carenze congenite di alcuni degli enzimi delle vie di interconversione, recupero e degradazione delle purine. La sintomatologia varia a seconda dai casi e può comprendere alterazioni neurologiche, miopatie, deficit immunitari e litiasi (Tab. 4.I).
Tab. 4.I. Classificazione dei difetti congeniti del metabolismo purinico
| Alterazione metabolica | principali sintomi |
| gotta familiare giovanile | iperuricemia, iperuricosuria |
| superattività di PRPP sintetasi | iperuricemia, iperuricosuria |
| deficit di ipoxantina fosforibosiltransferasi | sindrome di Lesch-Nyhan |
| deficit di adenina fosforibosiltransferasi | nefrolitiasi |
| xantinuria ereditaria | nefrolitiasi |
| deficit del cofattore per il molibdeno | grave encefalopatia |
| deficit di adenilosuccinato liasi | ritardo psicomotorio, epilessia |
| superattività di 5’-nucleotidasi | infezioni ricorrenti, epilessia |
| deficit di ecto-5’-nucleotidasi | immunodeficienza |
| deficit di adenosina deaminasi | immunodeficienza |
| deficit di nucleoside fosforilasi | immunodeficienza |
| deficit di mioadenilato deaminasi | miopatia |
Alterazioni del metabolismo purinico e dell’escrezione di acido urico possono portare a iperuricemia o a ipouricemia.
4.1.1. Iperuricemia
L’iperuricemia è classificata come primitiva quando non è conseguente a una malattia acquisita né è una manifestazione tardiva o secondaria di una alterazione morbosa di tipo diverso (Tab. 4.II). Questo disordine metabolico è generalmente a carattere familiare e, in circa il 25% dei pazienti, sembra essere legato ad un’aumentata sintesi delle purine dovuta a fattori diversi da caso a caso e solo in parte noti. In altri soggetti l’iperuricemia può essere dovuta ad una diminuita capacità del rene ad eliminare l’acido urico. L’aumento della concentrazione di acido urico nei liquidi biologici può determinare la precipitazione di questo catabolita nei tessuti con comparsa di tofi e l’insorgenza della gotta (caratterizzata da episodi ricorrenti di artrite acuta e dalla presenza di depositi uratici nelle articolazioni, nelle zone ad esse adiacenti e nel rene).
Tab. 4.II. Classificazione delle iperuricemie
| Primitive | |||
| aumentata sintesi purinica | |||
| diminuita escrezione renale | |||
| Secondarie | |||
| deficit enzimatici ereditari | |||
| deficit di ipoxantina fosforibosiltransferasi (s. di Lesch-Nyhan) | |||
| iperattività di PRPP sintetasi | |||
| deficit di glucosio 6-fosfatasi (glicogenosi I) | |||
| chetoacidosi (m. ad urine a sciroppo d’acero) | |||
| emopatie su base proliferativa | |||
| policitemie secondarie a: | |||
| cardiopatie congenite | |||
| affezioni broncopolmonari | |||
| carcinoma renale | |||
| leucemie | |||
| aumentata emocateresi | |||
| talassemia | |||
| anemia drepanocitica | |||
| ittero emolitico | |||
| emofilia | |||
| malattia di von Willebrand | |||
| saturnismo | |||
| aumentato consumo di alcool etilico | |||
| carico di fruttosio (intolleranza congenita) | |||
| farmaci | |||
Particolarmente alta è l’incidenza di iperuricemia nelle
emopatie su base proliferativa (policitemie secondarie a cardiopatie congenite,
ad affezioni broncopolmonari o a carcinoma renale, varie forme di leucemia
esclusa la linfatica, mieloma multiplo) oltre che nelle forme morbose
caratterizzate da intensa distruzione di elementi cellulari nucleati, come ad
esempio l’anemia drepanocitica (vedi
Par. 7.2.1a), la talassemia
(vedi Par.7.2.1b), l’ittero emolitico
(vedi Par. 7.1.3a), l’emofilia
e la malattia di von Willebrand
![]() (vedi
Par. 7.3.3a). Il controllo del
metabolismo dell’acido urico è essenziale nel corso della terapia della leucemia
mieloide cronica: la rapida lisi cellulare durante una crisi blastica può
infatti determinare un brusco aumento dell’uricemia con conseguente insorgenza
di un danno renale.
(vedi
Par. 7.3.3a). Il controllo del
metabolismo dell’acido urico è essenziale nel corso della terapia della leucemia
mieloide cronica: la rapida lisi cellulare durante una crisi blastica può
infatti determinare un brusco aumento dell’uricemia con conseguente insorgenza
di un danno renale.
Un’iperuricemia datante dall’infanzia, che può evolvere in
gotta dopo i dieci anni, è presente in soggetti affetti da glicogenosi tipo I
![]() ed è probabilmente causata sia da un aumentato
catabolismo dei nucleotidi purinici sia da un’inibizione della secrezione
dell’acido urico a livello dei tubuli renali, a sua volta conseguente
all’aumento in concentrazione dell’acido lattico nel sangue. Un’inibizione della
secrezione tubulare di acido urico attribuibile all’accumulo di chetoacidi
plasmatici sarebbe causa anche dell’iperuricemia nel corso della malattia con
urine a sciroppo d’acero
ed è probabilmente causata sia da un aumentato
catabolismo dei nucleotidi purinici sia da un’inibizione della secrezione
dell’acido urico a livello dei tubuli renali, a sua volta conseguente
all’aumento in concentrazione dell’acido lattico nel sangue. Un’inibizione della
secrezione tubulare di acido urico attribuibile all’accumulo di chetoacidi
plasmatici sarebbe causa anche dell’iperuricemia nel corso della malattia con
urine a sciroppo d’acero
![]() . Meccanismi simili a quelli descritti per
la glicogenosi tipo I sembrano essere alla base dell’iperuricemia osservabile a
seguito di somministrazione di fruttosio (specie in soggetti con intolleranza
congenita a questo zucchero
. Meccanismi simili a quelli descritti per
la glicogenosi tipo I sembrano essere alla base dell’iperuricemia osservabile a
seguito di somministrazione di fruttosio (specie in soggetti con intolleranza
congenita a questo zucchero
![]() ; vedi Fig. 4.5) o di
bevande alcoliche.
; vedi Fig. 4.5) o di
bevande alcoliche.
Grave forme di iperuricemia con precoce comparsa di tofi e
nefrolitiasi si osservano in pazienti con iperattività di PRPP sintetasi o con
sindrome di Lesch-Nyhan
![]() .
.
Numerosi farmaci possono determinare iperuricemia
probabilmente alterando la clearance renale dell’acido urico. Fra questi bisogna
ricordare i diuretici (tiazide, clorotiazide, acetozolamide, acido etacrinico,
furosemide), gli antitubercolari (pirazinamide, etambutolo), gli anestetici
generali (metossifluorano) ed altri ancora (salicilati a dosi basse
![]() , acido nicotinico, levodopa, agenti
citolitici, abuso di lassativi). L’iperuricemia
è stata riscontrata inoltre in casi di saturnismo.
, acido nicotinico, levodopa, agenti
citolitici, abuso di lassativi). L’iperuricemia
è stata riscontrata inoltre in casi di saturnismo.
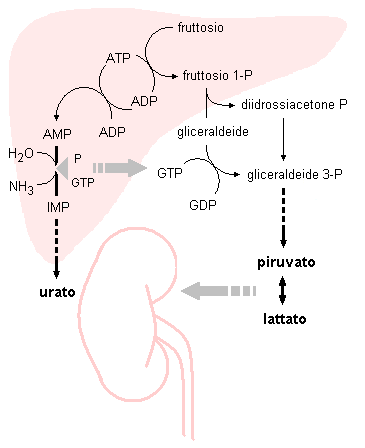
Fig 4.5. Schema ipotetico della patogenesi dell’iperuricemia da carico di fruttosio. Il metabolismo del fruttosio, che avviene principalmente nel fegato, porta ad un accumulo di acido lattico ed a un consumo di fosfato inorganico e di GTP (inibitori allosterici della AMP deaminasi). Ciò determina rispettivamente una inibizione della secrezione tubulare renale degli acidi organici e una attivazione del catabolismo dei nucleotidi adenilici. Entrambi questi fattori concorrono ad aumentare l’uricemia.
4.1.2. Ipouricemia
Si definisce ipouricemico un soggetto con una
concentrazione di acido urico nel siero, determinata con un metodo colorimetrico,
inferiore a 149 μM (2,5 mg/dL) se
maschio o a 125 μM (2,1 mg/dL) se
femmina (Tab. 4.III). L’ipouricemia è una condizione relativamente rara,
rilevabile nel 0.18% della popolazione, che spesso si trasmette come un
carattere autosomico recessivo (ipouricemia ereditaria renale
![]() ).
).
Altri difetti ereditari del trasporto renale dell’acido
urico, che possono dar luogo ad ipouricemia, fanno parte di sindromi più complesse
che coinvolgono un disturbo generalizzato del trasporto di membrana, come la
sindrome di Hartnup
![]() , la sindrome di Fanconi-DeToni-Debré
, la sindrome di Fanconi-DeToni-Debré
![]() e i disordini
metabolici correlati a quest’ultima sindrome (morbo di Wilson
e i disordini
metabolici correlati a quest’ultima sindrome (morbo di Wilson
![]() , cistinosi
, cistinosi
![]() , galattosemia
, galattosemia
![]() , intolleranza ereditaria al fruttosio
, intolleranza ereditaria al fruttosio
![]() ). Altre condizioni che
possono portare ad ipouricemia, inducendo la tubolopatia caratteristica della
sindrome di Fanconi-DeToni-Debré, sono alcune malattie neoplastiche (quali il
mieloma multiplo, il linfoma, il morbo di Hodgkin
). Altre condizioni che
possono portare ad ipouricemia, inducendo la tubolopatia caratteristica della
sindrome di Fanconi-DeToni-Debré, sono alcune malattie neoplastiche (quali il
mieloma multiplo, il linfoma, il morbo di Hodgkin
![]() e il carcinoma polmonare) ed
alcuni stati di intossicazione (avvelenamenti da metalli pesanti o derivati
delle tetracicline). Un’ipouricemia di origine metabolica può essere dovuta a
xantinuria ereditaria
e il carcinoma polmonare) ed
alcuni stati di intossicazione (avvelenamenti da metalli pesanti o derivati
delle tetracicline). Un’ipouricemia di origine metabolica può essere dovuta a
xantinuria ereditaria
![]() , a deficit di nucleoside fosforilasi purinica
, a deficit di nucleoside fosforilasi purinica
![]() o a deficit
combinato di xantina ossidasi, sulfito ossidasi e aldeide ossidasi
o a deficit
combinato di xantina ossidasi, sulfito ossidasi e aldeide ossidasi
![]() .
Un’ipouricemia acquisita di origine renale può essere presente in pazienti con
un aumento dei fluidi extracellulari (ad esempio, per una inappropriata increzione dell’ormone antidiuretico;
vedi Par. 10.1). Un’ipouricemia di natura iatrogena può
derivare dalla somministrazione di allopurinolo
.
Un’ipouricemia acquisita di origine renale può essere presente in pazienti con
un aumento dei fluidi extracellulari (ad esempio, per una inappropriata increzione dell’ormone antidiuretico;
vedi Par. 10.1). Un’ipouricemia di natura iatrogena può
derivare dalla somministrazione di allopurinolo
![]() in casi di gotta primitiva o
metabolica o per prevenire un’urolitiasi nel corso di un intervento terapeutico
aggressivo su neoplasie.
in casi di gotta primitiva o
metabolica o per prevenire un’urolitiasi nel corso di un intervento terapeutico
aggressivo su neoplasie.
|
Tab. 4.III. Classificazione delle ipouricemie |
|||
|
|
|||
| Diminuita formazione di acido urico | |||
| deficit metabolici | |||
| deficit di nucleoside fosforilasi purinica | |||
| xantinuria ereditaria | |||
| deficit del cofattore del molibdeno | |||
| ipouricemia iatrogena | |||
| somministrazione di allopurinolo | |||
| Aumentata escrezione renale | |||
| ipouricemia ereditaria renale | |||
| sindrome di Hartnup | |||
| sindrome di Fanconi-DeToni-Debrè | |||
| primitiva | |||
| secondaria a deficit metabolici | |||
| cistinosi | |||
| intolleranza ereditaria al fruttosio | |||
| galattosemia | |||
| morbo di Wilson | |||
| secondaria a malattie neoplastiche | |||
| mieloma multiplo | |||
| linfoma | |||
| morbo di Hodkin | |||
| carcinoma polmonare | |||
| secondaria ad avvelenamenti | |||
| metalli pesanti | |||
| tetracicline | |||
| Aumento dei fluidi extracellulari | |||
| iperincrezione dell’ormone antidiuretico | |||
|
|
|||
4.1.3. Metodi di determinazione
L’acido urico può essere dosato direttamente per mezzo di metodi chimici ed enzimatici o dopo essere stato purificato.
4.1.3a. Riduzione dell’acido fosfotungstico.
Il metodo si basa sulla riduzione dell’acido fosfotungstico in ambiente alcalino ad opera dell’acido urico con formazione di un composto colorato che assorbe a 700 nm. La procedura originale prevedeva la deproteinizzazione del campione mediante precipitazione (al calore o in presenza di acido tungstico, acido fosfotungstico o acido tricloroacetico) e l’isolamento dell’acido urico dal filtrato come sale d’argento prima di farlo reagire con l’acido fosfotungstico in una soluzione di carbonato di sodio. In alternativa, l’acido urico può essere isolato come sale di magnesio, ammonio o rame, la reazione può essere alcalinizzata con urea e cianuro (cosa che evita di dover isolare l’acido urico dal filtrato), l’agente ossidante può essere sostituito con acido arsenotungstico, acido arsenofosfotungstico, acido arsenomolibdico, ferrocianuro di potassio o acetato di uranile. Interferenze possono essere causate dalla presenza di sali ferrosi, acido ascorbico, glucosio, glutatione, cisteina, cistina, tirosina, triptofano o fenoli.
4.1.3b. Dosaggio enzimatico.
In presenza dell’enzima uricasi, l’acido urico è ossidato ad acido 5-idrossiurico che si decompone spontaneamente in allantoina.
![]()
Valori falsamente bassi possono essere ottenuti in presenza di
alte concentrazioni di xantina (in pazienti trattati con
allopurinolo
![]() ) per inibizione competitiva dell’uricasi. La
reazione può essere seguita spettrofotometricamente a 293 nm (in
ambiente alcalino) o a 283 nm (in ambiente acido) in quanto l’allantoina,
a differenza dell’acido urico, non assorbe a queste
lunghezze d’onda. Poiché le proteine assorbono notevolmente
nel vicino ultravioletto, è consigliabile usare un campione
preventivamente deproteinizzato.
) per inibizione competitiva dell’uricasi. La
reazione può essere seguita spettrofotometricamente a 293 nm (in
ambiente alcalino) o a 283 nm (in ambiente acido) in quanto l’allantoina,
a differenza dell’acido urico, non assorbe a queste
lunghezze d’onda. Poiché le proteine assorbono notevolmente
nel vicino ultravioletto, è consigliabile usare un campione
preventivamente deproteinizzato.
E’ possibile inoltre seguire il consumo di ossigeno nella reazione catalizzata dall’uricasi mediante un polarografo. Questo tecnica d’analisi, raramente adoperata, subisce interferenze da parte dell’allopurinolo, della xantina e dell’ipoxantina. Il perossido di idrogeno, prodotto durante l’ossidazione dell’acido urico, può essere dosato spettrofotometricamente in diversi modi (vedi Tab. IV).
| Tab.4.IV. Dosaggio del perossido d’idrogeno prodotto dalla reazione catalizzata dall’uricasi. | ||
|
|
||
| Reattivi | Enzima | Lunghezza d’onda (nm) |
|
|
||
| o-dianisidina | perossidasi | 530 |
|
3-metil-2-benzotiazolinone N,N-dimetilanilina |
perossidasi | 600 |
|
3,5-dicloro-2-idrossibenzensulfonato 4-aminofenazone |
perossidasi | 520 |
|
2,4,6-tribromofenolo 4-aminofenazone |
perossidasi | 492 |
|
etanolo NAD+ |
catalasi aldeide deidrogenasi |
340 |
|
|
||
La specificità dipende dal metodo di analisi. Il saggio utilizzante le reazioni catalizzate dalla catalasi e dall’aldeide deidrogenasi è il più frequentemente usato.

4.1.3c. Purificazione dell’analita.
L’acido urico è purificato mediante cromatografia liquida ad alta pressione con colonne a scambio ionico o in fase inversa utilizzando un’analizzatore spettrofotometrico (a 235 o 280 nm) o amperometrico. Questo metodo di analisi è sensibile e specifico ma raramente usato in laboratori di biochimica clinica. La separazione mediante cromatografia liquida ad alta pressione dei metaboliti intermedi nella via di biosintesi ed interconversione delle purine è particolarmente utile per la diagnosi biochimica degli errori congeniti del metabolismo. L’acido metilurico (metabolita della teofillina; vedi Par.13.3i) può interferire con il dosaggio dell’acido urico nelle urine in quanto è difficilmente separabile da questo con tecniche cromatografiche.
4.1.4. Preparazione del campione ed intervalli di riferimento
Plasma, siero e urine possono essere usati nella determinazione dell’acido urico. Può essere utile dosare l’acido urico nelle urine delle 24 ore. Nello studio di difetti metabolici ereditari è invece utile determinare il rapporto acido urico/creatinina nelle urine. E’ comunque consigliabile aggiungere 10 ml di NaOH (10 M) nel recipiente che dovrà servire per la raccolta dell’urine allo scopo di evitare una eventuale precipitazione dell’acido urico in ambiente acido. L’analita è stabile per circa tre giorni a temperatura ambiente, ammesso che si possa evitare una sua distruzione ad opera dei batteri. La corretta scelta di un valore di riferimento discriminante situazioni iper- e normouricemiche è ostacolata dal fatto che la distribuzione di frequenza dell’uricemia è spesso asimmetrica o bimodale ed è fortemente influenzata da fattori razziali e socioeconomici. Una migliore definizione di iperuricemia si basa sulla determinazione del limite di solubilità dell’acido urico (circa 6,8 mg/dL in una soluzione salina isotonica). Tenuto conto che circa 0,4 mg/dL di acido urico sono normalmente legati alle proteine plasmatiche si può presumere che l’acido urico sia prossimo alla precipitazione nei liquidi biologici a concentrazioni superiori a 7 mg/dL (416 µmol/L).
4.2. METABOLISMO PIRIMIDINICO
La sintesi dell’anello pirimidinico è catalizzata nel citoplasma della cellula da un complesso multienzimatico costituito dalla carbamilfosfato sintetasi II, dall’aspartato transcarbamilasi e dalla diidroorotasi. L’enzima carbamilfosfato sintetasi II forma il carbamilfosfato utilizzando glutamina, CO2 e due molecole di ATP attraverso una reazione analoga a quella catalizzata dall’enzima mitocondriale carbamilfosfato sintetasi I, che è coinvolto nel ciclo dell’urea (vedi Fig. 6.1). L’aspartato transcarbamilasi catalizza la formazione del carbamilaspartato dal carbamilfosfato e dall’aspartato, mentre la diidroorotasi completa la chiusura dell’anello pirimidinico formando l’acido diidroorotico.
Come è decritto nel Par. 6, anche l’enzima mitocondriale carbamilfosfato sintetasi I contribuisce in modo significativo alla sintesi epatica delle pirimidine giacché in condizioni normali circa un terzo del carbamilfosfato sintetizzato nel mitocondrio è esportato nel citoplasma ed è incorporato per circa l’80% nelle pirimidine.
L’ossidazione dell’acido diidroorotico ad acido orotico è catalizzata da una deidrogenasi specifica localizzata sulla superficie della membrana interna del mitocondrio. L’acido orotico può essere anche assunto con la dieta in quanto è contenuto nel latte bovino (circa 80 mg/L) e nei suoi derivati. L’acido orotico è convertito in UMP dalla UMP sintetasi, un enzima citosolico bifunzionale che si comporta sia come orotato fosforibosiltransferasi sia come orotato 5’-monofosfato decarbossilasi.
I nucleosidi pirimidinici monofosfati sono fosforilati a nucleosidi difosfati da due chinasi che utilizzano l’ATP e il dATP come donatore del gruppo fosforico e sono specifiche in un caso per il CMP, l’UMP e il dCMP e nell’altro caso per il dTMP e il dUMP. I nucleosidi difosfati sono convertiti in nucleosidi trifosfati dalla nucleoside difosfochinasi, un enzima che accetta come substrato sia i ribonucleosidi che i deossiribonucleosidi difosfati purinici e pirimidinici. La riduzione dei ribonucleosidi difosfati in deossinucleosidi difosfati avviene a spese della tioredoxina che, a sua volta, viene riconvertita nella forma ridotta dall’NADPH per mezzo della tioredoxina reduttasi. L’UTP è convertito in CTP in presenza di ATP, glutamina o NH3 nella reazione catalizzata dalla CTP sintetasi.
Le basi pirimidiniche non sono riutilizzate in quantità significativa nei tessuti umani in quanto le fosforilasi pirimidiniche vengono adoperate dall’organismo essenzialmente per la degradazione dei nucleosidi e non in direzione della loro sintesi (Fig. 4.6). Il riutilizzo dei nucleosidi pirimidinici è invece reso possibile per la presenza di uridina chinasi, deossicitidina chinasi e timidina chinasi. Tutti e tre gli enzimi fosforilano i nucleosidi pirimidinici nei corrispondenti nucleotidi a spese dell’ATP. La deossicitidina chinasi è specifica per un solo nucleoside, l’uridina chinasi accetta come substrato sia l’uridina che la citidina, e la timidina chinasi accetta come substrato sia la timidina che la deossiuridina.
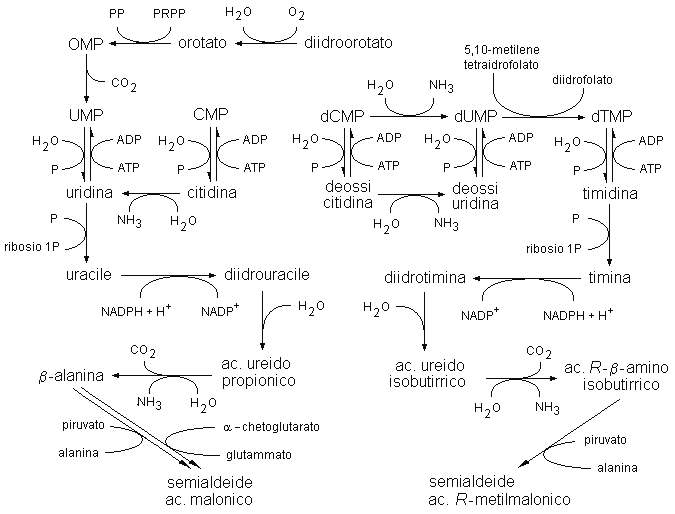
Figura 4.6. Vie di recupero e di degradazione delle pirimidine
I mononucleotidi pirimidinici sono idrolizzati nei corrispondenti nucleosidi dalle 5’-nucleotidasi. L’ulteriore degradazione dei nucleosidi avviene attraverso due distinte vie metaboliche che riguardano rispettivamente i ribonucleotidi ed i deossiribonucleotidi. Queste vie metaboliche prendono origine dall’uridina e dalla timidina e portano alla formazione di carbamil-β-aminoacidi e successivi prodotti di degradazione. La citidina è convertita in uridina dalla citidina deaminasi. La deossicitidina è convertita in timidina attraverso una catena di reazioni, che passa attraverso la deossiuridina o il dCMP, porta in sequenza alla formazione di dUMP, dTMP e timidina, e coinvolge gli enzimi deossicitidina deaminasi, deossicitidina monofosfato deaminasi, deossicitidina chinasi, timidina chinasi, timidilato sintetasi e 5’-nucleotidasi.
L’uridina e la timidina sono convertite nelle rispettive basi libere da due enzimi specifici, l’uridina fosforilasi e la timidina fosforilasi. Le successive reazioni sono catalizzate dalla diidropirimidina deidrogenasi, dalla diidropiramidasi e dalla β-ureidopropionasi, un enzima presente esclusivamente nel fegato. Per mezzo di questi enzimi l’uracile viene ridotto a diidrouracile, l’anello pirimidinico viene rotto con formazione dell’acido ureidopropionico e quest’ultimo viene degradato a β-alanina, NH3 e CO2. La timina è ridotta a diidrotimina, convertita ad acido ureidoisobutirrico ed infine degradata ad acido R-β-aminoisobutirrico, NH3 e CO2.
La β-alanina è presente allo stato libero in molti tessuti e nel plasma (generalmente ad una concentrazione inferiore a 14 μmol/L). La β-alanina è utilizzata nella sintesi della carnosina (N-β-alanil-istidina), un dipeptide presente nei muscoli scheletrici e nel cervello. La β-alanina può inoltre andare incontro a transaminazione con l’α-chetoglutarato o il piruvato, formando la semialdeide dell’acido malonico. Quest’ultimo composto subisce una serie complessa di trasformazioni biochimiche che portano alla formazione di acetil-CoA e di altri acil derivati.
L’acido R-β-aminoisobutirrico viene in parte escreto con le urine (10-235 μmol nelle 24 ore). L’acido R-β-aminoisobutirrico può inoltre formare, per transaminazione con il piruvato, la semialdeide dell’acido R-metilmalonico. La racemizzazione della forma R nella forma S permette a questo composto di utilizzare le reazioni enzimatiche, proprie del catabolismo della L-valina, che portano alla formazione di succinil-CoA.
4.2.1. Alterazioni metaboliche
Una diminuzione fino al 50% dell’escrezione urinaria di
acido orotico può essere riscontrata nei casi di digiuno prolungato. Più
numerose e frequenti sono le condizioni che portano ad un incremento
dell’escrezione urinaria di acido orotico. In Tab. 4.V sono elencati gli
errori congeniti che possono dare luogo a questo tipo di alterazione metabolica.
La presenza di grandi di quantità di acido orotico nelle urine (600 – 1500 mg al
giorno) con cristalluria ed ostruzione del tratto urinario può essere
riscontrata nella aciduria orotica tipo I caratterizzata da un difetto sia della
fosforibosilazione che della decarbossilazione dell’acido orotico. Estremamente
più rara è l’aciduria orotica tipo II con perdita dell’attività decarbossilasica,
ma mantenimento della capacità di fosforibosilare l’acido orotico a OMP. Di più
frequente riscontro sono i deficit degli enzimi del ciclo dell’urea (vedi
Par. 6.1c) e fra questi
il più comune è il deficit di ornitina transcarbamilasi che si manifesta con una
frequenza di 1:14000 e si trasmette come un carattere ereditario legato al
cromosoma X. Per quanto riguarda questo difetto metabolico devono essere
differenziati tre gruppi di pazienti: i maschi emizigoti con una grave
iperescrezione urinaria di acido orotico, le pazienti di sesso femminile con una
attività enzimatica residua variabile a seconda di come sono stati inattivati i
cromosomi X durante l’embriogenesi, e le pazienti eterozigote asintomatiche che
presentano solo un lievissimo aumento della escrezione urinaria di acido orotico.
Più raramente, un aumento di escrezione di acido orotico è dovuto ad un alterato
trasporto intramitocondriale dell’ornitina
![]() , ad un disordine del metabolismo della
lisina
, ad un disordine del metabolismo della
lisina
![]() o ad altri deficit enzimatici (deficit
combinato di glutammato formiminotransferasi e formimino tetraidrofolato
ciclodeaminasi o deficit di PRPP sintetasi).
o ad altri deficit enzimatici (deficit
combinato di glutammato formiminotransferasi e formimino tetraidrofolato
ciclodeaminasi o deficit di PRPP sintetasi).
| Tab. 4.V. Errori metabolici che portano ad una aumentata escrezione urinaria di acido orotico | |
|
|
|
| deficit degli enzimi del metabolismo pirimidinico | |
| orotato fosforibosiltransferasi / OMP decarbossilasi (aciduria orotica tipo I) | |
| OMP decarbossilasi (aciduria orotica tipo II) | |
| deficit degli enzimi del ciclo dell’urea | |
| ornitina transcarbamilasi | |
| argininosuccinato sintetasi (citrullinemia) | |
| argininosuccinato liasi (argininosuccinatemia) | |
| arginasi (argininemia) | |
| alterato trasporto intramitocondriale dell’ornitina | |
| sindrome da iperornitinemia-iperammoniemia-omocitrullinemia | |
| disordini del metabolismo della lisina | |
| intolleranza proteica lisinurica | |
| iperlisinemia periodica con iperammoniemia | |
| altri rarissimi deficit enzimatici | |
| glutammato formiminotransferasi / formimino tetraidrofolato ciclodeaminasi | |
| deficit di PRPP sintetasi | |
|
|
|
Un significativo aumento della escrezione urinaria di acido
orotico può essere indotto dalla somministrazione di 6-azauridina in pazienti
affetti da cancro o dalla somministrazione di allopurinolo o oxipurinolo
![]() , che
inibiscono l’attività della OMP decarbossilasi. Elevate concentrazioni di acido
orotico nelle urine sono state riscontrate negli alcolisti. Lievi o moderati
aumenti dell’aciduria orotica sono infine osservabili in pazienti affetti da
diabete, ipertensione, insufficienza cardiaca, infezioni cerebrali, tumori o
traumi.
, che
inibiscono l’attività della OMP decarbossilasi. Elevate concentrazioni di acido
orotico nelle urine sono state riscontrate negli alcolisti. Lievi o moderati
aumenti dell’aciduria orotica sono infine osservabili in pazienti affetti da
diabete, ipertensione, insufficienza cardiaca, infezioni cerebrali, tumori o
traumi.
4.2.2. Metodi di determinazione
L’acido orotico è dosabile direttamente nei liquidi biologici o dopo purificazione mediante cromatografia o elettroforesi.
4.2.2a. Dosaggio diretto dell’analita
L’acido orotico può essere convertito in acido barbiturico mediante brominazione e riduzione in presenza di acido ascorbico. L’acido barbiturico, a sua volta, è fatto reagire con la p-dimetilaminobenzaldeide per formare un prodotto di colore arancione che assorbe la luce a 480 nm. Questo metodo può subire l’interferenza di numerosi composti e dosa simultaneamente sia l’acido orotico che l’orotidina. Tuttavia, nei campioni d’urina solo l’istidina interferisce significativamente nell’analisi; per correggere questa interferenza è necessario allestire un controllo non brominato.
Un metodo enzimatico per il dosaggio dell’acido orotico si basa sull’utilizzo dell’orotato fosforibosiltransferasi e dell’orotato 5’-monofosfato decarbossilasi. La reazione può essere seguita direttamente a 295 nm. Un altro saggio enzimatico prevede l’utilizzo della diidroorotato deidrogenasi e la misura del prodotto di reazione a 282 nm.
Altri metodi di analisi diretta dell’acido orotico in campioni di urina prevedono l’utilizzo di uno spettrometro di massa (l’analita è identificato sul picco con m/z 155) o di uno strumento per la risonanza magnetica del protone (il segnale di risonanza è a 6,22 ppm). Entrambi i metodi sono di infrequente uso in laboratori d’analisi ad alta specializzazione.
4.2.2b. Purificazione dell’analita
La purificazione parziale dell’acido orotico in campioni d’urina può essere ottenuta mediante cromatografia su Dowex 1X2, amberlite CG 120 o gel di silice. L’assorbimento dell’eluato è seguito a 280 nm.
La purificazione dell’acido orotico mediante cromatografia liquida ad alta pressione su colonne in fase inversa è insoddisfacente poiché l’analita viene eluito troppo vicino al fronte d’analisi. Migliori risultati possono essere ottenuti con l’uso di due colonne cromatografiche in serie o attraverso la derivatizzazione dell’acido orotico nel suo estere metilico.
Un altro metodo d’analisi proposto per l’acido orotico prevede la trasformazione dell’analita nel suo trimetilsilil derivato e l’utilizzo di un gas cromatografo accoppiato ad uno spettrometro di massa.
Una eccellente purificazione dell’acido orotico da campioni biologici può essere ottenuta mediante l’utilizzo dell’elettroforesi capillare (Fig. 4.7). In un tampone alcalino ed in presenza di un sufficiente flusso elettroosmotico, tutti i composti presenti nel campione di urina migrano verso il catodo. In tali condizioni è possibile separare bene l’acido orotico dall’uracile e dall’acido urico, ma non si può escludere che eventuali altre sostanze possano rendere di difficile interpretazione il tracciato elettroforetico interferendo nell’analisi. In un tampone con un basso valore di pH, si ha un minore rischio di interferenze in quanto l’acido orotico migra verso l’anodo assieme ad un ristretto numero di composti con un sufficiente stato di ionizzazione.
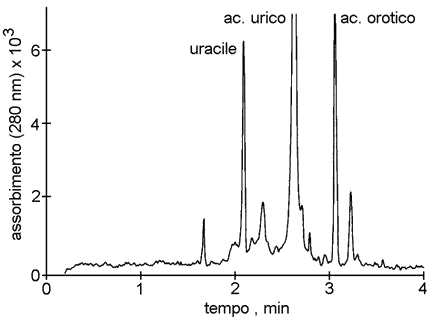
Fig. 4.7. Elettroferogramma di un campione di urine di un paziente con sindrome da iperornitinemia-iperammoniemia-omocitrullinemia. Il tracciato è stato ottenuto mediante elettroforesi capillare del campione in tampone borato (20 mmol/L, pH 9,2). Questa tecnica permette la determinazione contemporanea di acido orotico ed uracile.
4.2.3. Preparazione del campione ed intervalli di riferimento
La concentrazione dell’acido orotico nel plasma è normalmente inferiore a 0,5 μmol/L. Nel plasma di pazienti con errori del metabolismo pirimidinico o del ciclo dell’urea si possono trovare concentrazioni di acido orotico superiori a 60 μmol/L.
Poiché l’escrezione renale dell’acido orotico è molto efficiente, il dosaggio di questo composto viene eseguito di preferenza su campioni di urine. La distribuzione di frequenza dei livelli urinari di acido orotico nei soggetti sani è asimmetrica con una media di 1,13 μmol/mmol creatinina e una moda di 0,62 μmol/mmol creatinina. Nelle determinazioni eseguite mediante cromatografia liquida ad alta presione, l’intervallo di riferimento è di 0,26 - 3,20 μmol/mmol creatinina. Il livello dell’acido orotico urinario è più alto nei soggetti di sesso femminile (0,36 - 3,20 μmol/mmol creatinina) rispetto ai maschi (0,26 - 1,91 μmol/mmol creatinina). Valori più elevati sono riscontrabili nei neonati di 1 - 12 mesi (0,76 - 4,10 μmol/mmol creatinina).
Nei soggetti sani l’escrezione di acido orotico si mantiene sufficientemente costante nel tempo. Nei pazienti con deficit di ornitina transcarbamilasi si osserva invece un aumento della escrezione urinaria di acido orotico durante le prime ore della giornata, in conseguenza del catabolismo degli aminoacidi ingeriti durante la colazione dopo il digiuno notturno. Se il paziente ha una ammoniemia nei limiti della norma, il livello dell’acido orotico urinario può scendere a valori molto bassi nelle ore pomeridiane, impedendo di effettuare una diagnosi biochimica del difetto metabolico.
Per diagnosticare lo stato di eterozigoti in donne a
rischio per il deficit ereditario di ornitina transcarbamilasi, è stato proposto
di eseguire la determinazione dell’acido orotico urinario dopo un carico orale
di allopurinolo (300 mg)
![]() . Sebbene il valore predittivo del test sia buono, è
consigliabile dosare contemporaneamente nelle urine anche l’orotidina per
evitare errori interpretativi del dato analitico. Bisogna inoltre ricordare che,
nei bambini non in buone condizioni di salute, un aumento dell’escrezione
urinaria di acido orotico dopo carico di allopurinolo è di frequente riscontro
anche in assenza di uno specifico difetto metabolico congenito.
. Sebbene il valore predittivo del test sia buono, è
consigliabile dosare contemporaneamente nelle urine anche l’orotidina per
evitare errori interpretativi del dato analitico. Bisogna inoltre ricordare che,
nei bambini non in buone condizioni di salute, un aumento dell’escrezione
urinaria di acido orotico dopo carico di allopurinolo è di frequente riscontro
anche in assenza di uno specifico difetto metabolico congenito.
Recentemente è stato proposto di dosare l’escrezione urinaria di uracile al posto dell’acido orotico per ottenere un parametro maggiormente utile per lo studio biochimico del deficit di ornitina transcarbamilasi.
| aggiornamento: 02/10/13 |