|
|
Costantino Salerno |
|
|
Appunti di Biochimica Clinica |
|
|
13. TOSSICOLOGIA CLINICA
La tossicologia analizza gli effetti lesivi che sono esercitati sui tessuti dalle sostanze chimiche estranee (tossine) che vengono introdotte nell’organismo. Una volta che l’organismo è venuto in contatto con queste sostanze, esse possono andare incontro a svariati processi. Lo studio della "tossicocinetica" permette di conoscere le trasformazioni subite dalle tossine nell’organismo (nel caso di composti ad azione farmacologica viene più propriamente adoperato il termina "farmacocinetica"). Per questo tipo di studi, ha notevole importanza conoscere la via di somministrazione e la dose della tossina somministrata così come la durata dell’esposizione ad essa. Una volta entrata nel torrente circolatorio, la distribuzione della tossina nei diversi apparati, organi e tessuti dipende soprattutto dalle sue caratteristiche fisico-chimiche, compreso il grado di idro- o liposolubilità e di ionizzazione.
Lo studio dell’interazione fra la tossina ed i tessuti è indicato con il termine di "tossicodinamica". La comprensione della natura dell’interazione è essenziale per un approccio intelligente alla diagnosi e al trattamento dell’avvelenamento. Le modificazioni metaboliche subite dalla tossina possono alterarne le proprietà e causarne l’inattivazione o, viceversa, la trasformazione in un metabolita ancora più tossico. Il monitoraggio delle alterazioni dei parametri di laboratorio indotte dalle tossine è di fondamentale importanza per una terapia mirata.
13.1. DROGHE D’ABUSO
Con il termine "droga" si indica ogni tipo di sostanza capace di modificare l’umore, la percezione della realtà o il comportamento del soggetto che l’assume, innescando in misura variabile un fenomeno di tossicodipendenza. Per quanto riguarda il suo carattere "di abuso", questo implica un giudizio morale mutabile nei tempi e dipendente dal contesto socio-culturale e legislativo delle diverse comunità umane. Per tale motivo, nell’applicazione medica e forense e negli interventi socio-sanitari è al criterio legale che pragmaticamente più spesso ci si riferisce nel giudicare illecita una droga. Bisogna ricordare, infatti, che il diffondersi del consumo di tabacco fu inizialmente fortemente ostacolato nei paesi occidentali, l’uso e la vendita di alcolici furono proibiti negli Stati Uniti durante il proibizionismo, mentre l’assunzione voluttuaria di morfina e cocaina fu per lungo tempo tollerata in Europa, specie fra la fine dell’ottocento e i primi anni del novecento. D’altra parte, il consumo di nicotina, alcool etilico e caffeina è oggi considerato lecito e persino incentivato dalla propaganda pubblicitaria mentre quello della morfina e della cocaina è fortemente combattuto in Occidente; l’uso dell’hashish è tollerato mentre quello dell’alcool è severamente proibito in diversi paesi del Medio Oriente; il peyote è adoperato in rituali religiosi da alcune comunità autoctone della America Centro-Settentrionale e le foglie di coca sono da sempre masticate dagli Indios delle Ande.
In base alla loro attività farmacologica sul sistema nervoso centrale, le droghe d’uso illecito sono classificabili in:
1. psicolettiche (deprimenti il sistema
nervoso, ad es. barbiturici, benzodiazepine, oppiacei)
2. psicoanalettiche (psicostimolanti, ad es.
cocaina, amfetaminici)
3. psicodislettiche (allucinogeni, cannabinoidi).
Gli esempi riportati forniscono solo un quadro limitato delle
sostanze chimiche (in continuo aumento) oggi presenti sul mercato
clandestino o sul normale circuito di distribuzione
![]() . E’ da notare,
inoltre, che alcuni composti possono avere effetti farmacologici
diversi. Così, la cocaina ha un’azione deprimente sul
sistema nervoso centrale a dosi elevate e in seguito ad abuso
cronico, alcuni derivati amfetaminici si comportano come
allucinogeni, mentre i cannabinoidi provocano effetti tipici solo
in parte confrontabili con quelli delle droghe psicodislettiche.
. E’ da notare,
inoltre, che alcuni composti possono avere effetti farmacologici
diversi. Così, la cocaina ha un’azione deprimente sul
sistema nervoso centrale a dosi elevate e in seguito ad abuso
cronico, alcuni derivati amfetaminici si comportano come
allucinogeni, mentre i cannabinoidi provocano effetti tipici solo
in parte confrontabili con quelli delle droghe psicodislettiche.
Assai raramente gli stupefacenti vengono commercializzati allo stato puro sul mercato clandestino. L’aggiunta di sostanze di "taglio" ha diverse finalità: (1) aumentare il profitto diluendo il principio attivo con sostanze inerti, (2) potenziare o migliorare l’effetto della droga primaria, (3) ostacolare il riconoscimento chimico della sostanza illegale, (4) elevare la tossicità della miscela fino a renderla letale.
La presenza di sostanze di taglio nella droga di strada ha una serie di conseguenze di carattere tossicologico, sanitario e legale. Infatti, oltre agli effetti tossici ascrivibili direttamente all’uso della sostanza stupefacente, si possono frequentemente osservare alterazioni patologiche dovute ad adulteranti e diluenti, processi infettivi dovuti ad un non-rispetto delle norme di asepsi, lesioni locali dovute alle modalità di assunzione, nonché fenomeni tossici dovuti ad altre sostanze psicotrope assunte in associazione.
La ricerca biochimica e il dosaggio del composto stupefacente nei liquidi biologici o nelle polveri di strada può avere scopi diversi. Nel primo caso gli obiettivi principali sono (1) epidemiologico-statistici per stime sulla diffusione della droga, (2) tossicologici per l’individuazione della sostanza responsabile di una intossicazione negli interventi di pronto soccorso o per scopi medico-legali, (3) di accertamento di una avvenuta assunzione di droga per sancire eventuali provvedimenti amministrativi o penali, (4) di controllo del rispetto del "contratto terapeutico" stipulato dal tossicodipendente. Per i reperti provenienti dal traffico illecito, invece, la finalità è quella di quantificare la sostanza presente in essi per discriminarli sulla base della dose minima utilizzabile per uso personale. Mentre la competenza di valutare la presenza di sostanze stupefacenti nei liquidi biologici può essere delle USL, degli Istituti di Farmacologia e di Medicina Legale delle Università e dei Presidi Multizonali di Prevenzione, il compito di esprimersi sulla quantità di principio attivo presente nella droga di strada rimane più propriamente appannaggio degli organi di Polizia, degli Istituti Medico-Legali e dei Presidi Multizonali di Prevenzione.
13.1a. Barbiturici
I barbiturici sono dei derivati della malonilurea usati in terapia come anticonvulsivanti (barbiturici ad azione lunga ed intermedia), ipnotici (barbiturici ad azione breve) e anestetici locali (barbiturici ad azione ultrabreve) da soli o in associazione tra loro o con altri farmaci, come l’aspirina, l’efedrina, la codeina, la caffeina e la teofillina (Fig. 13.1).
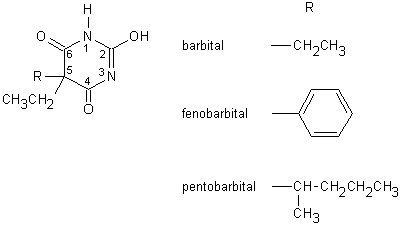
Fig. 13.1. Barbiturici
I barbiturici sono facilmente assorbiti dopo somministrazione orale (la loro biodisponibilità è pari al 90-100%). Essi vanno incontro a reazioni metaboliche comprendenti l’ossidazione dei sostituenti in posizione C5, la N-dealchilazione in posizione N1 e la distruzione dell’anello eterociclico. Questi metoboliti si formano in quantità variabile: ad esempio, il pentobarbital viene modificato per più del 95%, il fenobarbital per il 75%, mentre il barbital viene escreto quasi completamente immodificato. I farmaci modificati o allo stato nativo vengono lentamente escreti dal rene come glucoronidi o altri coniugati. Tracce di questi composti sono rinvenibili nelle urine fino a 16 giorni dall’assunzione.
13.1b. Benzodiazepine
Le benzodiazepine presentano un anello benzenico fuso con un
anello diazepinico a 7 atomi. Le più importanti contengono un
terzo anello benzenico come sostituente in posizione C5 (il
diazepam è, ad esempio, il 7-cloro-1-metil-5-fenil-1,4-benzodiazepin-2-one;
Fig. 13.2). La natura e la posizione di altri
sostituenti possono avere importanti effetti sull’azione
farmacologica della molecola
![]() . Sono stati finora sintetizzati
oltre 1000 composti di questo tipo. Quelli presenti sul mercato
si trovano principalmente sotto forma di capsule, compresse o
preparazioni iniettabili e sono usati in terapia come
tranquillanti, ipnotici, anticonvulsivanti e miorilassanti.
. Sono stati finora sintetizzati
oltre 1000 composti di questo tipo. Quelli presenti sul mercato
si trovano principalmente sotto forma di capsule, compresse o
preparazioni iniettabili e sono usati in terapia come
tranquillanti, ipnotici, anticonvulsivanti e miorilassanti.
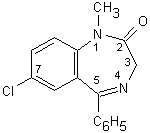
Fig. 13.2. Diazepam
Le benzodiazepine sono ben assorbite dopo somministrazione orale (la loro biodisponibilità varia dal 50 al 100% a seconda delle caratteristiche fisico-chimiche della molecola). In base alla loro lipofilicità, si accumulano più o meno rapidamente nel sistema nervoso centrale, nel tessuto adiposo e nei muscoli. Le benzodiazepine sono in gran parte metabolizzate dal fegato in composti che mantengono le proprietà farmacologiche, ma che generalmente vengono eliminati dall’organismo più lentamente rispetto alla molecola di partenza. Le trasformazioni chimiche a cui vanno incontro comprendono (1) la modificazione e/o la rimozione del sostituente in posizione N1, (2) l’eventuale idrossilazione in posizione C3 e (3) la coniugazione con acido glucuronico dei metaboliti idrossilati. L’escrezione è lenta e spesso i metaboliti sono rinvenibili nelle urine fino a 12 giorni dopo l’assunzione.
13.1c. Oppiacei
L’oppio è costituito dal lattice essiccato ottenuto per incisione delle capsule ancora verdi, ma a pieno sviluppo del Papaverum somniferum. Esso è costituito da una miscela complessa di zuccheri, proteine, lipidi, sostanze gommose e circa 40 alcaloidi biologicamente attivi, assommanti nel loro complesso a circa il 10-20% del peso totale del lattice. Gli alcaloidi dell’oppio sono divisi in due classi principali: gli alcaloidi isochinolinici (papaverina, narcotina; Fig. 13.3) e gli alcaloidi fenantrenici (morfina, codeina, tebaina; Fig. 13.4).
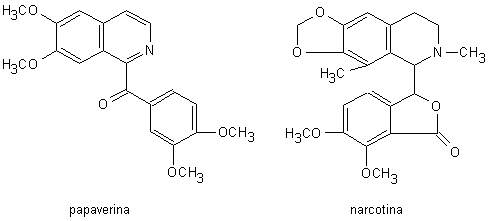
Fig. 13.3. Alcaloidi isochinolinici
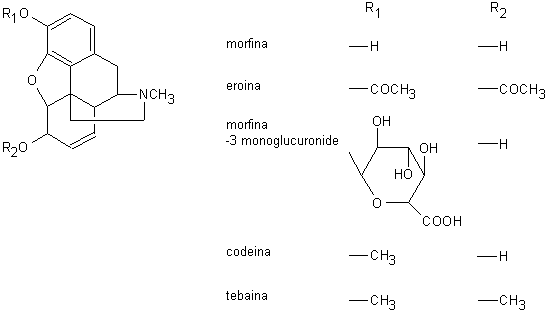
Fig. 13.4. Alcaloidi fenantrenici
L’attività stupefacente è presente solo negli alcaloidi a nucleo fenantrenico, pur presentando gli alcaloidi isochinolinici azioni farmacologiche importanti. La morfina è il principale alcaloide dell’oppio, essendo presente ad una concentrazione dell’8-14%. Gli alcaloidi dell’oppio sono in gran parte salificati dall’acido meconico (Fig. 13.5), che è presente ad una concertazione di circa il 15%.
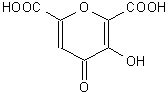
Fig. 13.5. Acido meconico
L’oppio che ha subito solo le manipolazioni necessarie al suo confezionamento è definito grezzo. L’oppio medicinale viene ottenuto essiccando l’oppio grezzo a temperatura moderata, riducendolo in polvere più o meno fine ed aggiustando il suo contenuto in morfina al 9,5-10,5% mediante aggiunta di lattosio, amido di riso o altri diluenti inerti. Preparazioni officinali sono l’estratto acquoso (20% di morfina), il laudano (1% di morfina) e la tintura idroalcolica (1% di morfina). La morfina viene comunemente estratta dall’oppio. Quella presente sul mercato, può essere di diversa qualità a seconda delle abitudini e della professionalità del laboratorio in cui è avvenuta la purificazione.
L’eroina (diacetilmorfina) è prodotta dalla morfina per acetilazione. I preparati commerciali possono contenere quantità anche notevoli di altri alcaloidi presenti nella morfina di partenza, non sufficientemente purificata. La presenza di acido meconico è indice di grossolana preparazione del prodotto. La presenza di 6-monoacetilmorfina (MAM) può essere dovuta ad una insufficiente acetilazione del prodotto o ad una successiva idrolisi dell’eroina, spesso per una eccessiva aggiunta di acido cloridrico durante il processo di salificazione. L’eroina è reperibile relativamente pura, diluita con zuccheri, o tagliata ulteriormente con sostanze che ne accentuano l’azione depressiva (chinino, barbiturici), aggiungono un’azione anestetica locale (procaina, lidocaina) o ne contrastano gli effetti secondari sgradevoli (amfetamina, caffeina, stricnina). A seconda della preparazione commerciale, l’eroina può essere classificata in eroina del Medio Oriente, eroina del Sud-Ovest Asiatico ed eroina del Sud-Est Asiatico (brown sugar). Quest’ultima esiste in due tipi: l’eroina da fumo contenente costantemente caffeina e spesso anche stricnina e l’eroina iniettabile, teoricamente pura.
Il metadone (Fig. 13.6), una sostanza ottenuta completamente per sintesi in laboratorio, pur avendo una struttura chimica diversa dalla morfina, viene spesso classificato fra le sostanze oppiaceo-simili in quanto possiede azioni farmacologiche analoghe ed è usato come sostituto della morfina nei trattamenti di disassuefazione.
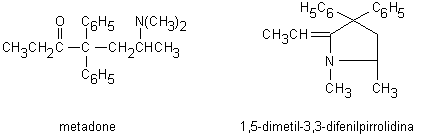
Fig. 13.6. Derivati del metadone
La morfina è facilmente assorbita dal tratto gastrointestinale, dalla mucosa nasale, dal polmone e per via subcutanea ed intramuscolare. La biodisponibilità di una preparazione orale è del 15-50% a causa della elevata inattivazione a livello della mucosa intestinale e del fegato, a cui arriva attraverso il sistema portale (effetto da primo passaggio). Nel plasma, circa un terzo del farmaco è legato alle proteine. La morfina si accumula rapidamente nei tessuti parenchimatosi, che lascia dopo breve tempo. Il più importante meccanismo di inattivazione metabolica della morfina è la sua coniugazione con l’acido glucuronico sull’ossidrile fenolico. L’eliminazione dall’organismo è rapida (l’emivita del farmaco è di circa 3 ore) ed avviene attraverso le urine e la bile. La presenza di un circolo enteroepatico della morfina libera e glucuronata giustifica il rinvenimento di piccole quantità di droga nelle urine anche alcuni giorni dopo l’ultima assunzione.
A causa della sua più elevata liposolubilità rispetto alla morfina, l’eroina attraversa più rapidamente la barriera ematoencefalica e permane per più tempo nel sistema nervoso centrale. La subitanea saturazione dei recettori cerebrali specifici provoca una sensazione particolarmente ricercata dal tossicodipendente e denominata flash. L’eroina viene quindi rapidamente idrolizzata dalle esterasi presenti nell’organismo a monoacetilmorfina e morfina, di cui segue il destino metabolico.
Il metadone si distingue dai composti precedenti per una maggiore stabilità e liposolubilità. Assunto per via orale non subisce un rilevante effetto da primo passaggio epatico e si concentra rapidamente nei tessuti lipofili ove mantiene a lungo concentrazioni elevate. La sua eliminazione dall’organismo è molto lenta (il tempo di dimezzamento è di circa 22 ore). Il principale metabolita del metadone è la 1,5-dimetil-3,3-difenilpirrolidina derivata dalla N-demetilazione e successive ciclizzazione della molecola (Fig. 13.6).
13.1d. Cocaina
La cocaina (l’estere metilico della benzoilecgonina; Fig. 13.7) è il principale alcaloide contenuto alla concentrazione di circa lo 0,1% nelle foglie della Erithroxylon coca. Queste vengono generalmente sottoposte ad essiccamento e trasformate in un omogenato (pasta di coca) ad elevato contenuto di principi attivi. La droga viene purificata mediante successive estrazioni in soluzione acquosa acida e solventi organici (dopo alcalinizzazione).
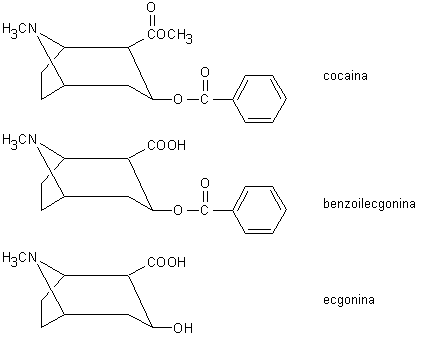
Fig. 13.7. Derivati della cocaina
La droga circola sui mercati internazionali ad un relativamente elevato grado di purezza. Occasionalmente, campioni di cocaina possono contenere grossi cristalli, a volte incolori (rock cocaine). Oltre che come sale, la cocaina può essere spacciata come base libera (crack) ottenuta per alcalinizzazione del prodotto ed evaporazione dell’acqua.
A livello di spaccio, la cocaina viene addizionata con diverse sostanze. Allo scopo di aumentare il peso del materiale commerciato, la cocaina viene addizionata a bicarbonato di sodio o a zuccheri. L’impiego di anestetici locali nel taglio ha lo scopo di surrogare la cocaina con molecole dotate di analoghe proprietà anestetico-locali e quindi difficilmente distinguibili ad un semplice esame organolettico. La presenza di acido acetilsalicilico, piramidone, caffeina, chinina e fenacetina è dovuta al fatto che questi prodotti, già miscelati, entrano nella composizione dei più comuni preparati ad azione antalgica. La procaina, oltre a conferire un maggior effetto anestetico, rende più difficile il riconoscimento della cocaina con i comuni reattivi, interferendo con eventuali accertamenti effettuati dalle forze dell’ordine.
La cocaina può essere miscelata a sostanze quali l’eroina e la morfina (speed ball ), che ne mitigano gli effetti eccitanti. Tali miscele sono particolarmente apprezzate nell’ambiente dell’uso illecito, soprattutto quando somministrate per via endovenosa.
La cocaina è ben assorbita per qualsiasi via di somministrazione. Per via endovenosa, la concentrazione plasmatica raggiunge immediatamente il suo massimo, diminuendo successivamente con un tempo di dimezzamento di circa 45-90 minuti. Per inalazione, il picco plasmatico viene raggiunto dopo circa un’ora, ma l’assorbimento è più rapido quando la droga viene assunta come base libera sotto forma di crack o miscelando nella pipa il cloridrato di cocaina con un alcalinizzante (ad esempio, bicarbonato di sodio). Ciò è dovuto da un lato alla maggiore volatilità (il punto di fusione scende da 198°C a 94°C, con conseguente minore suscettibilità del prodotto ad alterazioni termiche) e alla maggiore diffusibilità della base attraverso le membrane biologiche rispetto al cloridrato e, dall’altro lato, all’ampio sviluppo superficiale disponibile a livello degli alveoli polmonari.
Dopo l’assorbimento, la cocaina è degradata da esterasi plasmatiche e, in un secondo tempo, da enzimi microsomiali epatici. I metaboliti principali sono la benzoilecgonina, che si forma per idrolisi del legame estere e liberazione di metanolo, e la ecgonina, che deriva dalla precedente a seguito della liberazione di acido benzoico. La ecgonina può venire a sua volta demetilata a norecgonina.
La quota di farmaco e di metaboliti escreti con le urine varia con il pH. A pH urinario acido è escreta una maggiore quantità di cocaina immodificata. A pH urinario alcalino aumenta la quantità di ecgonina escreta. La escrezione della benzoilecgonina non risente invece del pH a causa della natura anfotera di questo composto.
13.1e. Amfetaminici
Questo gruppo di sostanze comprende l’amfetamina (metilfenetilamina) e i suoi derivati per sostituzioni sull’anello benzenico (Fig. 13.8). Queste sostanze possono provenire dai normali circuiti di distribuzione dei prodotti farmaceutici o essere sintetizzate clandestinamente. In quest’ultimo caso, il prodotto contiene spesso impurezze ed adulteranti. Sul mercato illecito, gli amfetaminici (chiamati in gergo speed) vengono generalmente tagliati al 40% con zuccheri, solfato di magnesio, glutammato di sodio o altri farmaci (caffeina, efedrina, procaina, antipirina, fenazone).
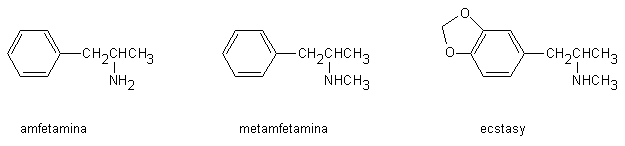
Fig. 13.8. Derivati dell’amfetamina
L’amfetamina e la metamfetamina (suo prodotto di metilazione) sono generalmente commercializzate sotto forma di sali (di colore giallo-brunastro a causa delle impurezze presenti) più stabili rispetto alle basi libere. La metamfetamina è venduta talora in soluzione acquosa (gold fish). La base libera della metamfetamina in forma cristallina (ice o shaboo) viene generalmente fumata o inalata dopo riscaldamento.
I derivati dell’amfetamina con sostituzioni sull’anello
benzenico, mai registrati e commercializzati come farmaci, sono
disponibili come basi libere (sotto forma di oli) o come sali.
Molti di questi derivati producono effetti farmacologici analoghi
alle sostanze psichedeliche (Par. 13.1f) ed
hanno talora un’attività più di cento volte superiore a
quella della mescalina, a cui sono chimicamente simili
![]() . Alcuni prodotti, che è
possibile rinvenire sul mercato clandestino, sono l’STP (Serenità,
Tranquillità e Pace; 2,5-dimetossi-4-metilamfetamina), il love
drug (3,4-metilendiossi-amfetamina) e l’ecstasy (3,4-metilendiossi-metamfetamina). La combinazione di ecstasy
e viagra
. Alcuni prodotti, che è
possibile rinvenire sul mercato clandestino, sono l’STP (Serenità,
Tranquillità e Pace; 2,5-dimetossi-4-metilamfetamina), il love
drug (3,4-metilendiossi-amfetamina) e l’ecstasy (3,4-metilendiossi-metamfetamina). La combinazione di ecstasy
e viagra
![]() prende
il nome di
sextasy sul mercato clandestino.
prende
il nome di
sextasy sul mercato clandestino.
Le amfetamine sono somministrabili per via orale. Esse superano facilmente la barriera ematoencefalica accumulandosi nel sistema nervoso centrale. Il loro metabolismo comporta una deaminazione ossidativa con formazione di derivati dell’acido benzenico e, dopo coniugazione con glicina, dell’acido ippurico. Le amine secondarie sono preventivamente demetilate ad amine primarie, prima di andare incontro alle trasformazioni descritte. Reazioni secondarie includono l’idrossilazione sull’anello aromatico e l’N-ossidazione (per i derivati dell’idrossilamina). Tutti questi composti sono coniugati con acido solforico o glucuronico.
L’escrezione urinaria dipende fortemente dal pH, così che la quantità eliminata per tale via può variare, nelle 24 ore, dal 75% (in urine acidificate) al 1% (in urine alcalinizzate).
13.1f. Allucinogeni
Gli allucinogeni sono un gruppo di sostanze, chimicamente differenti tra loro, capaci di indurre uno stato allucinatorio e, a volte, delirante caratterizzato da una iper-recettività agli stimoli esterni con un diminuito controllo della situazione vissuta dal soggetto, che ha spesso la sensazione in parte di diventare esso stesso spettatore passivo di quanto accade e in parte di partecipare sensitivamente all’esperienza (viaggio).
Queste sostanze possono essere classificate, in base allo loro origine, in:
1. allucinogeni di derivazione vegetale (mescalina,
psilocibina, salvinorina A)
2. allucinogeni di semisintesi (LSD)
3. allucinogeni di sintesi (fenciclidina, ketamina).
La mescalina (3,4,5-trimetossifenetilamina; Fig. 13.9) è il principale alcaloide contenuto in un piccolo
cactus senza spine e molto succulento, che è chiamato peyote (Lophophora
williamsii) ed è endemico in alcune regioni del Messico.
Anche altre specie di cactus della regione andina sono capaci di
produrre mescalina; esse sono note come San Pedro in Perù
e Aguacolla in Equador
![]() .
.
La psilocibina (4-fosforiloxi-N,N-dimetiltriptamina; magic
mushroom; Fig. 13.9) è il principale
alcaloide contenuto in numerose specie di funghi del genere Psilocybe
(le più importanti dal punto di vista dell’abuso sono
le P. mexicana, semilanceata e cubensis)
![]() .
.
La salvinorina A (Fig. 13.9) è contenuta nella Salvia divinorum, una pianta della famiglia delle Lamiaceae simile alla menta, che è coltivata dagli amerindi mazatechi nel sud del Messico. La salvinorina A è un potente allucinogeno, che è attivo a dosi inferiori a 100 μg.
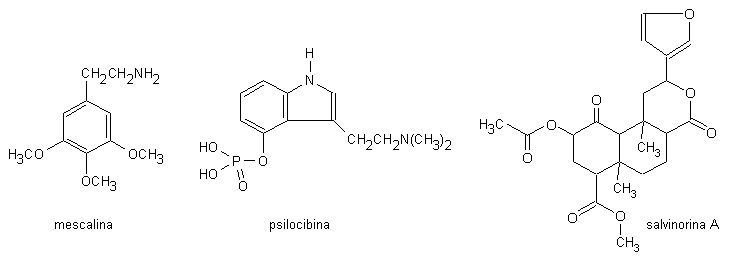
Fig. 13.9. Allucinogeni di derivazione vegetale
Un altro potentissimo allocinogeno è la
dietilamide dell’acido lisergico (LSD;
Fig. 13.10). Questo composto è sintetizzato a partire dall’acido
lisergico
![]() , che viene generalmente ottenuto da alcaloidi (ergometrina ed altri)
estratti dalle forme vegetative (sclerozi) della segale cornuta (Claviceps
purpurea), un fungo parassita delle piante di segale. Sul mercato
clandestino è venduto in compresse (microdots) da
circa 50-100 μg, assorbito su carta bibula o su
zollette di zucchero oppure incluso in una matrice di gelatina (window glass).
, che viene generalmente ottenuto da alcaloidi (ergometrina ed altri)
estratti dalle forme vegetative (sclerozi) della segale cornuta (Claviceps
purpurea), un fungo parassita delle piante di segale. Sul mercato
clandestino è venduto in compresse (microdots) da
circa 50-100 μg, assorbito su carta bibula o su
zollette di zucchero oppure incluso in una matrice di gelatina (window glass).
La fenciclidina (Fig. 13.10) è un anestetico generale che non ha superato la fase di sperimentazione clinica sull’uomo per i notevoli disturbi a cui andavano incontro i pazienti al momento del risveglio e che è stato essenzialmente usato in medicina veterinaria. Il composto, che può essere facilmente sintetizzato in laboratori clandestini ed immesso sul mercato a basso costo e in un vario grado di purezza, è reperibile sotto forma di pastiglie, capsule, polveri o soluzioni iniettabili o mescolato a tabacco, majurana o thè ed è chiamato in gergo angel dust, horse tranquilizer o in altri modi.
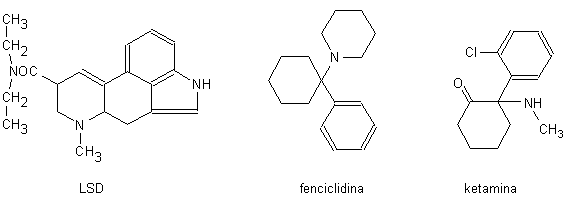
Fig. 13.10. Allucinogeni di semisintesi e di sintesi
Un analogo della fenciclidina è la ketamina, 2-(o-clorofenil)-2-(metilamino)-cicloesanone (Fig. 13.10). Questa sostanza è adoperata in veterinaria, in ortopedia e in cardiochirurgia durante l’inserzione di pace-makers. Se usata a scopo voluttuario (Special K) ha un effetto brevissimo, circa 10 minuti, ma lascia l’assuntore in uno stato depressivo grave costellato di allucinazioni e di forme di dissociazione.
La mescalina, la psilocibina e l’LSD sono generalmente assunte per via orale. La fenciclidina può essere invece presa tramite varie vie (orale, intranasale, rettale, vaginale, endovenosa o attraverso il fumo).
Tutti questi allucinogeni vanno incontro in buona parte a trasformazioni metaboliche. La mescalina è trasformata per circa il 70% nell’inattivo acido 3,4,5,-trimetossifenilacetico. La psilocibina è estesamente defosforilata a psilocina, che tuttavia mantiene l’attività biologica. Circa il 99% dell’LSD è inattivato in 2-ossidietilamina e in 13- e 14-idrossidietilamide. La fenciclidina subisce per l’85% processi di idrossilazione e N-dealchilazione con formazione di derivati idrossilici con nucleo cicloesanico o piperidinico e di acido aminopentanoico. Il tempo di dimezzamento varia fra le 3 ore (LSD) e le 16 ore (fenciclidina).
13.1g. Cannabinoidi.
I cannabinoidi sono un gruppo di composti (circa
60) a 21 atomi di carbonio aventi una struttura dibenzopiranica
![]() e presenti unicamente
nelle piante di canapa (Cannabis sativa). I principali
cannabinoidi presenti sono il Δ9-
e il Δ8-tetraidrocannabinolo
(THC) e i due isomeri dell’acido Δ9-tetraidrocannabinolico (rispettivamente con il
gruppo carbossilico in posizione 2 e 4 dell’anello fenolico;
Fig. 13.11). I due acidi organici sono
instabili al calore e tendono a decarbossilarsi in Δ9-THC. Quest’ultimo si
trasforma a sua volta, soprattutto in ambiente acido, nel meno
attivo Δ8-THC. Altri
cannabinoidi privi di azione stupefacente sono il cannabinolo (CBN)
e il cannabidiolo (CBD).
e presenti unicamente
nelle piante di canapa (Cannabis sativa). I principali
cannabinoidi presenti sono il Δ9-
e il Δ8-tetraidrocannabinolo
(THC) e i due isomeri dell’acido Δ9-tetraidrocannabinolico (rispettivamente con il
gruppo carbossilico in posizione 2 e 4 dell’anello fenolico;
Fig. 13.11). I due acidi organici sono
instabili al calore e tendono a decarbossilarsi in Δ9-THC. Quest’ultimo si
trasforma a sua volta, soprattutto in ambiente acido, nel meno
attivo Δ8-THC. Altri
cannabinoidi privi di azione stupefacente sono il cannabinolo (CBN)
e il cannabidiolo (CBD).
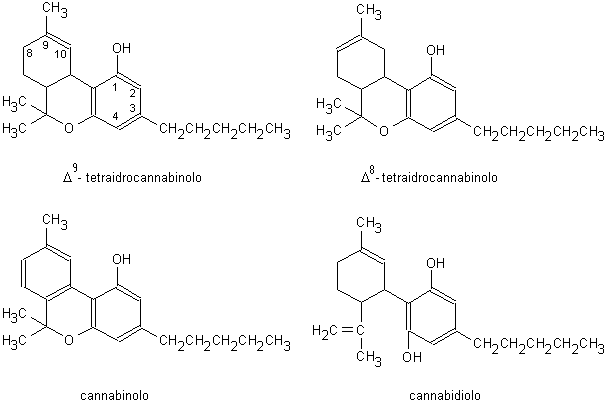
Fig. 13.11. Cannabinoidi
La canapa da droga (fenotipo I) non è facilmente differenziabile da quella tessile (fenotipo II). A livello europeo si è convenuto di considerare "canapa indiana", cioè canapa con potenzialità stupefacente, quelle piante con un contenuto di THC superiore allo 0,5%. Un altro parametro importante è il rapporto fenotipo, dato dal valore numerico ottenuto utilizzando le concentrazioni percentuali dei rispettivi cannabinoidi nella formula ([THC]+[CBN])/[CBN], che deve essere superiore all’unità nella canapa da droga. Le piante da droga, inoltre, sono in media un po’ più basse (specialmente gli esemplari femminili) e presentano un fusto ramificato fin dalle base e foglie dentellate con segmenti più sottili e più chiari rispetto alla varietà tessile. Tuttavia queste caratteristiche morfologiche sono fortemente influenzate da fattori ambientali e possono essere facilmente messe in evidenza solo confrontando i due fenotipi nelle medesime condizioni di coltivazione.
In commercio sono reperibili diversi preparati a base di canapa indiana. La marijuana è ottenuta mescolando varie parti della pianta, ha un contenuto medio di THC dello 0,5-1,5% ed è di solito fumata da sola o mescolata al tabacco. Il bhang è equivalente alla marijuana per preparazione, ma viene generalmente bevuto come decotto. Il ganja è un omogenato costituito da resina e dalle foglie proveniente dalle sommità fiorite della pianta femminile, ha un contenuto di THC pari al 2-4% ed è commercializzato in bastoncini o tavolette di diverso colore ("nero" del Pakistan e della Afganistan, "verde" di Tijuana, "oro" di Acapulco). L’hashish è costituito dalla resina relativamente pura estratta dalle sommità fiorite della pianta ed ha un contenuto di THC pari al 3-7%. L’olio di hashish è un concentrato ottenuto per percolazione dal materiale vegetale con il contenuto di THC del 20-40%, che è spesso aggiunto alle sigarette di maijuana per aumentare il loro contenuto di droga.
Il Δ9-THC è assorbito dal tratto gastrointestinale in maniera lenta ed irregolare, mentre per via inalatoria, quando è assunto tramite il fumo, il suo assorbimento è rapido (la biodisponibilità è di circa il 18% della dose assunta).
La droga viene ossidata in 11-idrossi-Δ9-THC e 8-idrossi-Δ9-THC (che sono biologicamente attivi) e in una serie di altri derivati idrossilati inattivi. Tutti questi composti sono coniugati in proporzioni variabili con acido glucuronico. La via principale di escrezione è rappresentata dalle feci. Il tempo di dimezzamento della droga assunta è di 14-38 ore. I metaboliti sono rinvenibili nelle urine anche dopo 12-15 giorni dall’assunzione di una singola dose.
13.1.1. Metodi di dosaggio
Nel dosaggio delle droghe d’abuso vengono generalmente distinti tre livelli di analisi: i test speditivi, i test di screening e i test di conferma.
13.1.1a. Test speditivi
Sono detti speditivi quei dosaggi che non richiedono strumentazioni sofisticate, sono eseguibili anche da personale non specializzato e servono di aiuto nell’orientare l’analisi successiva quando non è noto quale sia il composto presente nel campione da esaminare. Questi metodi si basano sull’uso di reagenti chimici relativamente specifici capaci di dar luogo, in presenza dell’analita, a prodotti colorati o fluorescenti o a precipitati facilmente identificabili ad occhio (Tab. 13.I). I metodi più comunemente usati impiegano capsule o piastre di vetro o, più comunemente, di porcellana bianca (per facilitare la percezione del colore che si sviluppa con la reazione) o provette di vetro. Riguardo al campione da esaminare, l’urina e il siero vengono adoperati senza alcun pretrattamento, per le polveri si utilizzano pochi granelli e per le compresse, i materiali vegetali o altri materiali resinosi si preleva con una spatola o un bisturi una piccola quantità che si spezzetta fino a ridurla in polvere.
Nell’interpretazione dei risultati è necessario tenere presente che solo il colore indicato per ogni singolo saggio deve essere interpretato come risultato positivo, intendendo con ciò la possibile presenza della sostanza per cui il metodo è proposto e non la prova definitiva della sua identificazione, che sarà ottenuta mediante tecniche più sofisticate.
|
Tab. 13.I. Test speditivi |
|||
| Reazione | Colore | Specificità | |
| ac. picrico | precipitato giallo arancio | alcaloidi | |
| cristalli aghiformi gialli | cocaina | ||
| Beam (potassa alcolica) | viola | canapa indiana | |
| cloruro ferrico | rosso | ac. meconico | |
| Dille-Koppanyi (acetato di Co, isopropilamina) | rosso porpora | barbiturici | |
| Dragendorff (iodobismutito) | precipitato arancio-bruno | alcaloidi | |
| Duquenois-Levis (vanillina, acetaldeide, HCl, cloroformio) | viola | canapa | |
| Ehrlich (p-DMBA, metanolo, ac. fosforico) | blu-viola | psilocibina, LSD | |
| rosso | fenciclidina | ||
| Fast blu B in cloroformio e soda | rosso | canapa | |
| ferricianuro di potassio | fluorescente | oppiacei | |
| Marquis (formaldeide e ac. solforico) | rosso-viola | oppioacei | |
| arancio | mescalina, psilocibina, amfetaminici | ||
| verde-bruno | psilocina | ||
| Nickolls (etanolo, HCl) | rosa | canapa | |
| permanganato di potassio | lamine rosa-violacee | cocaina | |
| sali di Co in etanolo e LiOH | blu | barbiturici | |
| Scott (polireazione) | tiocianato di Co | blu | cocaina |
| + HCl | rosa | cocaina | |
| + cloroformio | blu | cocaina | |
| soluzione iodo-iodurata | precipitato rosso-bruno | alcaloidi | |
| vanillina in ac. solforico a caldo | colori viranti col raffreddamento | barbiturici | |
| Vitali-Morris (ac. nitrico e potassa) | giallo | benzodiazepina | |
| Zimmermann (nitrobenzene e potassa) | rosso-porpora | benzodiazepina | |
13.1.1b. Test di screening
I test di screening hanno lo scopo di evitare nel maggior numero dei casi il ricorso a tecniche più sofisticate e costose in termini di tempo e di denaro (test di conferma), permettendo inoltre l’esecuzione rapida di analisi per un intero pannello di sostanze stupefacenti - cosa ancora più apprezzata quando si presume che il tossicodipendente abbia assunto più sostanze contemporaneamente così da rendere impraticabile l’analisi attraverso metodiche cromatografiche.
La tecnica alla base di questi metodi è generalmente quella immunochimica fondata sulla competizione, per i siti leganti di un anticorpo specifico, fra l’antigene "marcato", appositamente aggiunto in quantità nota alla miscela di reazione, e l’antigene "non marcato", presente nel campione da esaminare.
La specificità del saggio dipende dal tipo di anticorpo usato che deve essere privo di reattività verso sostanze a struttura chimica simile, ma in cui è assente l’azione stupefacente. Tuttavia, a volte, un certo grado di reattività crociata può essere utile al fine di poter evidenziare l’intera famiglia farmacologica invece della singola molecola (come, ad esempio, nel caso delle benzodiazepine)
La sensibilità è molto influenzata dal tipo di marcatore usato. E’ importante che il test di screening sia altamente sensibile e che la sensibilità sia dichiarata dal produttore. Infatti, mentre un falso positivo può essere comunque scoperto dal test di conferma, il falso negativo viene perso, non essendo più sottoposto ad analisi. Ciò può comportare per il laboratorio serie responsabilità, anche di carattere legale.
La rapidità dell’analisi dipende dalla fase scelta,
eterogenea (RIA) od omogenea (FPIA, EMIT
![]() ). Le due ultime metodiche
sono più rapide e facilmente automatizzabili, essendo in grado
di fornire i risultati in meno di mezz’ora. Nei sistemi
EMIT, gli enzimi usati come marcatori possono risentire anche
fortemente di variazioni di pH e temperatura e della presenza di
adulteranti. Più elevate prestazioni in termini di rapidà,
sensibilità e accuratezza sono fornite dai metodi FPIA.
). Le due ultime metodiche
sono più rapide e facilmente automatizzabili, essendo in grado
di fornire i risultati in meno di mezz’ora. Nei sistemi
EMIT, gli enzimi usati come marcatori possono risentire anche
fortemente di variazioni di pH e temperatura e della presenza di
adulteranti. Più elevate prestazioni in termini di rapidà,
sensibilità e accuratezza sono fornite dai metodi FPIA.
13.1.1c.Test di conferma
Questi metodi consentono la separazione, l’identificazione e, il più delle volte, il dosaggio quantitativo dell’analita in questione. Essi si basano su tecniche di cromatografia su strato sottile, di cromotografia liquida ad alta pressione (HPLC) e di gas cromatografia.
La cromatografia su strato sottile presenta spesso degli svantaggi rispetto gli altri metodi, dovuti alla modesta sensibilità (intorno al mg) e alla scarsa riproducibilità, in parte superati con la comparsa sul mercato di nuove fasi stazionarie e di sistemi automatizzati d’analisi. L’identificazione della sostanza avviene generalmente tramite l’uso di raggi UV o di reattivi chimici specifici.
I rilevatori utilizzati in HPLC sono quelli spettrofotometrici (con sensibilità di 0,01-1 μg), spettrofluorimetrici (con sensibilità di circa 1 ng) o elettrochimici (amperometrici con sensibilità di circa 50 pg e coulometrici con sensibilità di 10-20 ng). L’accoppiamento con la spettrofotometria di massa permette sensibilità di 1-5 pg. I rilevatori generalmente usati in gas cromatografia sono a ionizzazione fiamma, a cattura d’elettroni o a fotoionizzazione.
Qualunque sia il metodo cromatografico usato, generalmente è necessario procedere in via preliminare all’estrazione della sostanza in esame dalla matrice biologica mediante opportuno solvente. E’ questa forse la fase più critica di tutto il processo cromatografico: infatti, se la metodica impiegata non garantisce una elevata efficienza - sia in termini di riproducibilità che di selettività - viene vanificato l’utilizzo di strumentazioni anche altamente sofisticate. Spesso è inoltre necessario l’impiego di tecniche di derivatizzazione allo scopo di modificare chimicamente l’analita e renderlo più idoneo alla separazione cromatografica.
13.1.2. Preparazione del campione e valori di riferimento
Un elemento fondamentale è quello della realizzazione di una adeguata catena di custodia del campione. Nel caso delle urine, il campione deve essere raccolto in locali idonei privi di lavandino per evitare diluizioni o sofisticazioni, il soggetto non deve avere abiti capaci di contenere eventuali campioni di altri soggetti e la raccolta stessa deve essere effettuata in presenza di persona responsabile per accertarne l’appartenenza al soggetto. Una volta ottenuto il campione (almeno 60 mL), ne deve essere controllata la temperatura, il peso specifico e il colore per verificare che si tratti veramente di urina. Successivamente bisogna dividerlo in due aliquote, una per analisi e l’altra da conservare in frigorifero per ulteriori accertamenti.
La scelta della metodica e dell’opportuno valore di soglia dipende dal tipo di valutazione e dalla finalità dell’esame. Laddove c’è la necessità di discriminare un assuntore da uno spacciatore con l’accertamento della presenza della sostanza nelle urine, è utile slittare la soglia verso livelli bassi per non rischiare di avere un risultato falsamente negativo, perché ciò avrebbe una ricaduta più nociva per il soggetto in esame, catalogabile in questo caso come spacciatore anche se in possesso di quantità pari o inferiore alla dose media giornaliera. Nel caso della valutazione di idoneità a svolgere mansioni o nel monitoraggio di pazienti inseriti in un programma di disassuefazione, la scelta della soglia deve orientarsi, invece, verso livelli alti, sufficientemente garantisti. Bisogna ricordare infine che la concentrazione urinaria di un determinato metabolita non è direttamente correlabile alla quantità di droga assunta, ma dipende anche dalla clearance renale del composto in esame, dalla sua farmacocinetica e dal tempo intercorso fra l’assunzione e l’accertamento (Tab. 13.II).
|
Tab. 13.II. Droghe d’abuso |
||||
| nome | dose
tipica |
emivita |
clearance |
%
immodificata |
| pentobarbital | 50-200 |
15-48 |
0,3-0,5 |
1 |
| fenobarbital | 50-200 |
48-120 |
0,06 |
20-35 |
| diazepan | 5-30 |
20-50 |
0,3-0,5 |
<1 |
| morfina | 5-10 |
2-4 |
15-20 |
<10 |
| eroina | 5-10 |
1-1,5 |
15-20 |
<1 |
| metadone | 40-100 |
15-60 |
1-2 |
5-50 |
| cocaina | 50-100 |
2-5 |
25-45 |
<10 |
| amfetamina |
5-10 |
4-24 |
20 |
|
| mescalina | 250-350 |
6 |
50-60 |
|
| psilocibina | 20 |
|||
| LSD | 0,03-0,2 |
3-4 |
1 |
|
| fenciclidina | 15-20 |
7-16 |
30-50 |
|
| ketamina |
50-200 |
3-4 |
16-22 |
2-5 |
| cannabinoidi | 14-38 |
<1 |
||
13.2. ALCOOL ETILICO
L’alcool etilico è presente in bevande ad ampia
diffusione nei normali circuiti di distribuzione dei prodotti
alimentari. Esso è ingerito, nei paesi occidentali, in modo
saltuario o abituale da più del 90% degli individui adulti,
dando luogo a quadri di
intossicazione acuta e cronica (interessanti
principalmente il sistema nervoso centrale e il fegato) dovuti ad
una azione citotossica sia diretta sia mediata dai suoi prodotti
catabolici. Inoltre, le bevande alcoliche spesso sono assunte assieme ad altre
sostanze potenzialmente dannose
![]() o esse stesse contengono
composti tossici di derivazione vegetale o provenienti dai recipienti in cui è
avvenuta la fermentazione (alcoli a basso peso molecolare, aldeidi, fenoli,
terpeni, tannini, metalli pesanti, etc.
o esse stesse contengono
composti tossici di derivazione vegetale o provenienti dai recipienti in cui è
avvenuta la fermentazione (alcoli a basso peso molecolare, aldeidi, fenoli,
terpeni, tannini, metalli pesanti, etc.
![]() ).
).
L’assunzione dell’alcool etilico avviene
generalmente per via orale, raramente attraverso i vapori. L’alcool
è assorbito in gran parte dalla porzione prossimale del piccolo
intestino, in modesta quantità dallo stomaco e dal grosso
intestino e in piccolissima quantità dalla mucosa della bocca e
dell’esofago. La velocità di assorbimento aumenta con il
rapido svuotamento gastrico, con la diluizione (è massima alla
concentrazione del 20%) e in presenza di anidride carbonica (ad
esempio, nel caso di ingestione di spumanti), mentre diminuisce
quando l’alcool è ingerito assieme ad altri alimenti. In
quest’ultima circostanza, una frazione dell’alcool
introdotto in dosi modeste viene ossidata nello stomaco dell’alcool
deidrogenasi e quindi non entra nella circolazione sistemica
![]() .
.
Il 2-10% dell’etanolo assorbito è escreto come tale
attraverso i polmoni, l’urina e il sudore. La quota
rimanente viene metabolizzata, principalmente dal fegato, tramite
l’ossidazione ad acetaldeide e, in minima parte, tramite la
glucuronazione, la solfatazione o l’esterificazione con
acidi grassi. L’ossidazione dell’alcool etilico è
operata dall’alcool deidrogenasi (responsabile per circa l’80%
del processo catabolico), dalla catalasi e dal sistema
microsomiale di ossidazione dell’etanolo (MEOS). Questi tre
sistemi differiscono per i cofattori e/o i substrati delle
reazioni: l’alcool deidrogenasi richiede il NAD+,
la catalasi il perossido d’idrogeno e il MEOS l’ossigeno
molecolare e l’NADPH. L’acetaldeide viene quindi
ossidata dall’aldeide deidrogenasi (NAD-dipendente ) ad
acido acetico
![]() , che sarà
a sua volta ossidato con produzione di anidride carbonica ed
acqua o convertito in altri composti, fra cui gli acidi grassi.
Un soggetto adulto generalmente è capace di metabolizzare 10-20
ml (8-16 g) di etanolo all’ora.
, che sarà
a sua volta ossidato con produzione di anidride carbonica ed
acqua o convertito in altri composti, fra cui gli acidi grassi.
Un soggetto adulto generalmente è capace di metabolizzare 10-20
ml (8-16 g) di etanolo all’ora.
La determinazione del livello di alcolemia è frequentemente richiesta nei reparti di pronto soccorso in quanto numerosi stati morbosi (incluso il coma) possono essere causati dall’abuso di sostanze alcoliche. In tali casi, l’analisi deve essere rapida per indirizzare l’intervento terapeutico e sufficientemente accurata per i risvolti medico-legali connessi alla diagnosi. A volte è anche necessario individuare il tipo di alcool ingerito (generalmente etanolo, talora metanolo, isopropanolo o glicol etilenico). Valutazioni semiquantitative del grado di ebbrezza alcolica possono essere richieste dagli organi di polizia o nel controllo degli utenti della strada.
13.2.1. Metodi di dosaggio
L’alcool etilico in fase gassosa reagisce con agenti fortemente ossidanti come il bicromato, il permanganato o l’acido osmico. La concomitante riduzione dell’agente ossidante determina un cambiamento di colore della miscela di reazione. Un altro metodo rapido, ma poco specifico, per la determinazione dell’alcolemia si basa sul cambiamento delle proprietà colligative del siero, evidenziate dall’abbassamento del punto di congelamento.
I metodi enzimatici per la determinazione del livello alcolemico sfruttano la reazione catalizzata dall’alcool deidrogenasi e la misura spettrofotometrica o fluorimetrica dell’NADH prodotto. Infine, l’alcool etilico può essere separato in gas-cromatografia e misurato mediante ionizzazione a fiamma.
Altri parametri, che possono essere utilizzati per
evidenziare un consumo smodato di alcool, sono l’aumento del livello ematico di
CDT (Carbohydrate Deficient Transferrin
![]() ) e di alcuni enzimi
epatici, aspartato ed alanina aminotransferasi e γ-glutamiltranspeptidasi
) e di alcuni enzimi
epatici, aspartato ed alanina aminotransferasi e γ-glutamiltranspeptidasi
![]() , specie se questo
aumento è associato a iperuricemia (vedi Par. 4.1.1)
e macrocitosi (vedi Par. 7.1.3a).
Nel caso che sia necessario verificare una tossicodipendenza pregressa o
monitorare pazienti che stanno disintossicandosi oppure eseguire indagini
medico-legali su cadavere, si può ricorrere al dosaggio dell’etil glucuronide e
dell’etil solfato
, specie se questo
aumento è associato a iperuricemia (vedi Par. 4.1.1)
e macrocitosi (vedi Par. 7.1.3a).
Nel caso che sia necessario verificare una tossicodipendenza pregressa o
monitorare pazienti che stanno disintossicandosi oppure eseguire indagini
medico-legali su cadavere, si può ricorrere al dosaggio dell’etil glucuronide e
dell’etil solfato
![]() nel sangue, nell’urina
(fino dopo 78 ore) o nei capelli (fino dopo 60 giorni).
nel sangue, nell’urina
(fino dopo 78 ore) o nei capelli (fino dopo 60 giorni).
13.2.2. Preparazione del campione e valori di riferimento
Nel caso di prelievo di sangue, bisogna evitare di disinfettare la cute con soluzioni alcoliche per non alterare il campione (si usano soluzioni contenenti perossido di idrogeno). Gli anticoagulanti non interferiscono con il dosaggio dell’analita. Poichè l’etanolo è meno concentrato nelle emazie (il 16% in meno rispetto al plasma), le analisi sul sangue intero danno valori più bassi rispetto a quelle su plasma o siero.
La concentrazione alcolica nelle urine è maggiore di circa il 25% rispetto al plasma. Tuttavia, a causa delle difficoltà o del ritardo nella raccolta, le urine sono meno frequentemente utilizzate per l’analisi. L’aria espirata e la saliva sono usate per dosaggi estemporanei in soggetti che si sospetta essere in stato di ebbrezza, in quanto non richiedono procedure invasive per il prelievo.
Bisogna prestare particolare attenzione nella conservazione
del campione prelevato, specie nelle indagini medico legali. I campioni
devono essere conservati ben tappati e, preferibilmente, in
frigorifero. Per prevenire la crescita batterica, si può
aggiungere ad esempio dell’azide sodica (5 mg/mL). L’interpretazione del dato analitico ai fini legali
dipende dalle leggi dei singoli paesi: generalmente un soggetto
è considerato intossicato quando l’alcolemia è superiore
a 1 g/L (22 mM). Valori superiori a 3 g/L sono generalmente
associati a coma
![]() .
.
13.3. MONITORAGGIO TERAPEUTICO DEI FARMACI
Il monitoraggio terapeutico dei farmaci si basa sul presupposto che questo tipo di indagine possa fornire informazioni utili per eseguire eventuali adeguamenti posologici. Per questo motivo, in generale, non è appropriato determinare il livello di un farmaco se si dispone di buoni indicatori endogeni dell’effetto farmacologico, come ad esempio la misura della pressione sanguigna in corso di terapia antipertensiva, la glicemia in pazienti sottoposti a trattamento con ipoglicemizzanti orali, la colesterolemia in pazienti trattati con farmaci ipocolesterolemizzanti oppure la valutazione degli indici di emocoagulazione dopo somministrazione di eparina o altri anticoagulanti.
Bisogna inoltre sottolineare che la determinazione della concentrazione plasmatica del farmaco è solo una parte della procedura di monitoraggio terapeutico, che non può prescindere dalla valutazione del quadro clinico complessivo del paziente. La regola da seguire è di prendersi cura in primo luogo del paziente e non del livello ematico delle sostanze somministrate. A volte è infatti possibile ottenere un controllo soddisfacente della sintomatologia con concentrazioni del farmaco inferiori a quella compresa nell’intervallo terapeutico e, in questo caso, non bisogna ovviamente fare alcun tentativo di aumentare la posologia. D’altra parte, se non si ottiene l’effetto desiderato, ciò può essere dovuto a fattori che prescindono dalla concentrazione ematica raggiunta, come ad esempio fenomeni di idiosincrasia individuale, spiazzamento dai siti di legame o d’azione per competizione farmacologica, induzione di sistemi epatici di metabolizzazione o alterazione dei meccanismi di assorbimento o di escrezione renale.
Queste considerazioni riducono il numero dei farmaci per i quali esiste un unanime consenso sul fatto che il dosaggio quantitativo sia terapeuticamente utile. I farmaci da sottoporre a monitoraggio devono generalmente presentare una o più delle seguenti caratteristiche:
1. un ristretto intervallo terapeutico, cioè concentrazioni
tossiche solo lievemente più alte di quelle terapeutiche;
2. sintomi di tossicità facilmente confondibili con quelli della
malattia sottostante;
3. capacità di alterare il proprio metabolismo mediante
induzione enzimatica o sviluppo di insufficienza renale;
4. assorbimento variabile con scarsa correlazione fra dose somministrata
e livello ematico raggiunto.
Inoltre, possono essere candidati ad un monitoraggio terapeutico, indipendentemente dal tipo di farmaco assunto, i pazienti molto giovani, molto anziani, con compromissione dell’assorbimento o della funzione epatica o renale, oppure con alterazioni genetiche tali da determinare importanti variazioni metaboliche. Un monitoraggio terapeutico può essere altresì indicato nei casi in cui il legame del farmaco alle proteine è alterato per la concomitante assunzione di altri farmaci o per l’abnorme livello delle proteine plasmatiche.
Può essere infine utile, anche se economicamente svantaggioso, monitorare un farmaco per verificarne la effettiva assunzione da parte del paziente (compliance). Questo può essere un problema soprattutto nel caso dei farmaci che sono somministrati una sola volta al giorno, poiché una mancata assunzione ha un effetto maggiore sul loro equilibrio dinamico. In altri casi può diventare invece un problema l’eccessiva compliance, che si può riscontrare nel caso di bambini, figli di genitori iperansiosi, o negli anziani che assumono molte medicine confondendone la posologia.
13.3a. Aminoglicosidici
La gentamicina e gli altri aminoglicosidici sono antibiotici che possono agire in modo sinergico ai β-lattamici (pennicillina) e che molto spesso risultano efficaci per combattere infezioni sostenute da germi anaerobi resistenti ad altri antibiotici. Sono farmaci idrosolubili ed ampiamente confinati al compartimento extracellulare con una emivita di 1-8 ore. La concentrazione plasmatica massima accettabile per la gentamicina è di 10 mg/mL (nel picco di assunzione) e di 2 mg/mL durante il trattamento. Tutti gli aminoglicosidici sono dei farmaci poco maneggevoli in quanto operano in un ristretto intervallo terapeutico e possono determinare danni all’udito ed ai reni in caso di sovradosaggio.
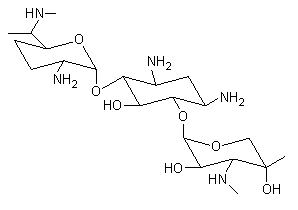
Fig. 13.12. Aminoglicosidici (gentamicina)
13.3b. Carbamazepina
La carbamazepina è un anticonvulsivante utilizzato nella terapia dell’epilessia. Si lega alle proteine plasmatiche, ma in modo variabile, e il suo dosaggio nella saliva a fini di monitoraggio non si è dimostrato di alcuna utilità. Viene metabolizzata principalmente nel fegato e trasformata nel 10,11-epossido che è ugualmente attivo, ma raramente dosato di routine. E’ un farmaco ad emivita breve (5-15 ore nei bambini). La carbamazepina è efficace a concentrazioni plasmatiche di 20-40 μmol/L. In circa il 30% dei pazienti compaiono tuttavia un gran numero di sintomi tossici cronici, tra cui una dermatite, anomalie ematologiche e segni di sofferenza epatica. Con valori superiori a 45 μmol/L, si manifestano nistagmo, atassia e sonnolenza.
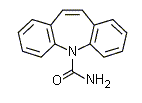
Fig. 13.13. Carbamazepina
13.3c. Ciclosporina
La ciclosporina è un peptide ciclico che trova un importante impiego come immunosoppressore nella chirurgia dei trapianti. Subisce complesse modificazioni metaboliche che la trasformano in numerosi derivati, molti dei quali ancora biologicamente attivi. Il dosaggio viene eseguito su sangue intero mediante cromatografia liquida ad alta pressione, che è specifica per il farmaco originale, o mediante metodi radioimmunologici, che possono essere specifici o aspecifici.
Durante il trattamento con ciclosporina, bisogna evitare sia i danni derivanti da un sottodosaggio del farmaco, che possono portare al rigetto dell’organo trapiantato, sia i danni da sovradosaggio a causa dell’elevata nefrotossicità di questa sostanza. Ciò assume particolarmente importanza in caso di trapianto renale in quanto non vi è ancora consenso unanime su quale sia l’intervallo terapeutico ottimale per la ciclosporina. Nella maggioranza dei casi i limiti inferiori e superiori si trovano entro l’intervallo di 100-200 μg/L.
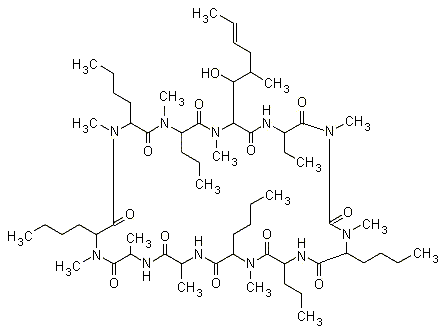
Fig. 13.14. Ciclosporina
13.3d. Difenilidantoina
La difenilidantoina è un farmaco anticonvulsivante comunemente usato nella terapia antiepilettica. Ha un buon assorbimento, una lunga emivita e viene metabolizzato prevalentemente dal fegato, che lo ossida. Tuttavia, la sua rimozione enzimatica è saturabile e il livello, al quale ciò accade, presenta una forte variabilità interindividuale. Inoltre, il farmaco può indurre la produzione di enzimi che promuovono il suo catabolismo, determinando una riduzione della sua concentrazione plasmatica. Alterazioni della clearance della difenilidantoina possono derivare dall’interazione con altri farmaci, soprattutto con il valproato.
Una volta avvenuta la saturazione della via metabolica che porta alla rimozione del farmaco, la concentrazione di difenilidantoina aumenta in modo esponenziale e ciò può accadere anche a livelli compresi nell’intervallo terapeutico, per cui piccoli aumenti della dose possono determinare cospicui incrementi della concentrazione plasmatica del farmaco. Gli adeguamenti individuali della posologia richiedono un attento monitoraggio del farmaco. La sua concentrazione plasmatica è comunemente mantenuta tra 40 e 80 μmol/L; livelli inferiori sono efficaci nel controllo di forme lievi di epilessia, mentre taluni pazienti necessitano di livelli anche superiori (90 μmol/L). Dosaggi eccessivi determinano depressione acuta del sistema nervoso centrale.
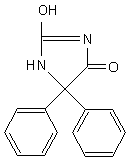
Fig. 13.17. Difenilidantoina
13.3e. Digossina
La digossina è un farmaco del gruppo della digitale che agisce inibendo la pompa sodio/potassio di membrana, soprattutto a livello miocardico dove esplica una azione inotropa positiva. Ha una emivita lunga (20-60 ore) e di solito viene somministrata una volta al giorno. Viene eliminata prevalentemente attraverso il rene e per questo motivo una riduzione del filtrato glomerulare può provocare una intossicazione da farmaco con bradicardia ed altre anomalie del ritmo cardiaco.
I pazienti sottoposti a terapia prolungata con digossina vanno incontro a fenomeni di adattamento. Per tale motivo, l’interpretazione dei valori plasmatici del farmaco è diversa a seconda che la terapia sia stata istituita da poco o da molto tempo. Effetti tossici si manifestano più probabilmente a concentrazioni superiori a 3 nmol/L. A qualunque livello del farmaco, la tossicità è tuttavia inversamente proporzionale alla concentrazione di potassio e, in presenza di ipopotassiemia, come quella indotta da alcuni diuretici, la tossicità può manifestarsi a valori di digossinemia attorno a 1,5 nmol/L.
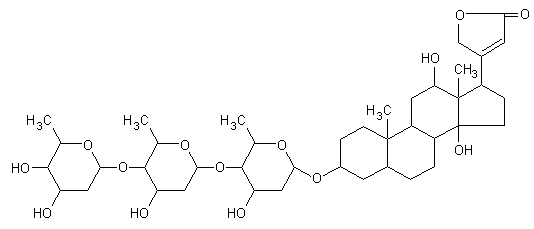
Fig. 13.15. Digossina
13.3f. Fenobarbitale
Il fenobarbitale è un barbiturico anticovulsivante (vedi Par. 13.1a) sia come tale, sia dopo essere stato trasformato in primidone, suo principale metabolita. Ha una emivita relativamente lunga e viene eliminato sia per via renale che per via epatica. Il metabolismo del fenobarbitale può variare notevolmente da individuo ad individuo in quanto il farmaco induce la sintesi epatica di enzimi microsomiali detossificanti. Per questo motivo non esistono limiti ben definiti per la concentrazione plasmatica di fenobarbitale (nella maggior parte dei pazienti ben controllati questa si aggira fra 80 e 160 μmol/L). I sintomi associati a dosaggi eccessivi vanno dalla sonnolenza al coma.
13.3g. Litio
Il litio viene somministrato per bocca sotto forma di carbonato nel trattamento della mania e della depressione bipolare. Ha una emivita di 20-40 ore e viene eliminato eclusivamente per via renale. I diuretici tiazidici possono aumentare la concentrazione plasmatica di litio, mentre il bicarbonato di sodio e lo spirolattone la riducono.
I livelli sierici di litio devono essere mantenuti tra 0,3 e 1,2 mmol/L in un campione prelevato a distanza di 12 ore dalla somministrazione. Raddoppiando il dosaggio si può arrivare ad una progressiva tossicità ed una concentrazione sierica superiore a 3,0 mmol/L è potenzialmente fatale. Livelli di litio non molto superiori a 1,2 mmol/L possono indurre nefrotossicità acuta ed i pazienti, che sviluppano diarrea o problemi renali, devono essere sottoposti a controlli regolari della litiemia. Un altro effetto collaterale è l’ipotiroidismo che, nei casi in cui non è possibile sospendere la terapia, deve essere trattato con tiroxina (vedi Par. 10.4).
13.3h. Metotrexato
Il metotrexato è un chemioterapico utilizzato, come antineoplastico, ad alti
dosaggi per via endovenosa per un periodo di 6-12 ore e, come antipsoriasico
![]() , a
basso dosaggio (15 mg). Il farmaco viene metabolizzato a 7-idrossi-metotrexato
ed eliminato per via renale.
, a
basso dosaggio (15 mg). Il farmaco viene metabolizzato a 7-idrossi-metotrexato
ed eliminato per via renale.
Il metotrexato è un inibitore del metabolismo dell’acido folico e, utilizzato ad alti dosaggi per un breve periodo, riesce ad eliminare le cellule neoplastiche in rapida mitosi, risparmiando quelle a crescita più lenta. Al termine dell’infusione, la concentrazione di metotrexato si riduce rapidamente. Se ciò non avviene o se la concentrazione plasmatica durante l’infusione è troppo elevata, deve essere somministrato al paziente l’acido folinico come fonte di folati. A tutti i dosaggi il farmaco risulta epatotossico; inoltre, sia il farmaco originario che il suo metabolita sono nefrotossici a causa della loro bassa solubilità.
La determinazione della concentrazione di metotrexato può essere richiesta per permettere l’aggiustamento della velocità d’infusione (la concentrazione ad equilibrio deve essere circa 100 μmol/L) o per controllare l’effettiva riduzione dei livelli del farmaco quando l’infusione viene sospesa.
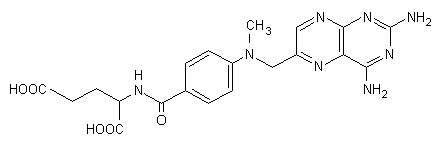
Fig. 13.16. Metotrexato
13.3i. Teofillina
La teofillina è un broncodilatatore utilizzato nella terapia dell’asma, ma è dotato di molti altri effetti collaterali (in particolare stimola la gittata cardiaca e determina la dilatazione dei vasi periferici). E’ somministrata per bocca come base libera o sotto forma salina oppure per endovena come sale idrosolubile (teofillina etilendiamina). Le preparazioni a lento rilascio permettono di fare assumere il farmaco ad intervalli di 12-24 ore.
La teofillina ha una breve emivita (3-13 ore) ed è metabolizzata in gran parte nel fegato attraverso il sistema dei citocromi P450: nel neonato (sotto i 6 mesi) viene prevalentemente trasformata in caffeina, sopra i sei mesi d’età e nell’adulto viene convertita in una varietà di cataboliti, tra cui la 3-metilxantina. Il metabolismo della teofillina è soggetto ad induzione da parte di altri farmaci, come la difenilidantoina. Inoltre, la teofillina mostra una velocità di escrezione dose-dipendente. Il farmaco ha una stretta finestra terapeutica (55-100 μmol/L nell’adulto, 25-80 μmol/L nel neonato). Sovradosaggi possono portare ad una grave forma di intossicazione, a volte fatale.
Fig. 13.18. Teofillina
13.3.1. Metodi di dosaggio
I metodi utilizzati nel dosaggio terapeutico dei farmaci comprendono tecniche immunologiche (isotopiche o ottiche), cromatografiche (liquide o su strato sottile) e gas-cromatografiche. La scelta della metodica da usare dipende da vari fattori: la richiesta di un servizio d’urgenza, la necessità di misurare più sostanze o più metaboliti in un medesimo campione, il costo e la durata dell’esame.
13.3.2. Preparazione del campione e valori di riferimento
Per la maggior parte delle analisi è possibile usare sia il siero che il plasma, comunque con l’avvertenza di evitare l’emolisi in quanto i farmaci possono concentrarsi negli eritrociti. Per la determinazione della ciclosporina è necessario adoperare il sangue intero. L’urina è di scarso valore nella valutazione quantitativa dei farmaci, ma l’analisi qualitativa può essere un modo per controllare la presenza di eventuali sostanze interferenti.
L’utilizzo di campioni di saliva può essere utile per la determinazione della quota libera del farmaco o per evitare il prelievo venoso nei bambini. La saliva è prodotta da tre diverse ghiandole e il suo pH può variare notevolmente, a seconda dello stimolo utilizzato. Ciò può avere un effetto sulla concentrazione salivare dei farmaci scarsamente oleosi. Il migliore stimolo è la masticazione di una banda elastica pulita.
Il momento migliore per effettuare il prelievo è quello immediatamente precedente una successiva somministrazione del farmaco. Fanno eccezione i farmaci aminoglicosidici, per i quali si è dimostrato che i livelli di picco danno luogo ad un indice indipendente di tossicità. Nel caso della digossina, il campione non dovrebbe essere prelevato entro 6 ore dalla somministrazione, poiché l’assorbimento di questa sostanza e il picco di distribuzione possono essere notevolmente elevati.
E’ opportuno prestare attenzione a possibili contaminazioni che possono derivare, ad esempio, dalla somministrazione di lidocaina o di altri anestetici a pazienti particolarmente agitati prima del prelievo e bisogna altresì avere l’accortezza di non raccogliere il campione di sangue dallo stesso braccio su cui è eventualmente inserita l’agocannula per una infusione. E’ infine opportuno limitare l’utilizzo di lacci emostatici nel caso che si debbano dosare farmaci con una alta affinità per le proteine (come ad esempio la difenilidantoina).
Anche se esiste un "intervallo terapeutico ematico" grossolano per tutti i farmaci comunemente sottoposti a monitoraggio, la concentrazione ottimale deve essere valutata nel paziente in base alla risposta individuale. Una volta individuato questo parametro, le eventuali variazioni della risposta sono riportate alla dose e alla conseguente concentrazione plasmatica del farmaco. Per la maggior parte dei farmaci vi è una relazione relativamente lineare tra dose somministrata e concentrazione plasmatica all’equilibrio dinamico; una eccezione a questa regola è rappresentata dalla difenilidantoina.
| aggiornamento: 03/10/14 |